Слайд 1ПРАВИЛА ГОСУДАРСТВЕННОЙ РЕГИСТРАЦИИ МЕДИЦИНСКИХ ИЗДЕЛИЙ СТРУКТУРА И СОДЕРЖАНИЕ Постановления правительства РФ (№ 1416, № 930, № 670)
Слайд 2Государственная регистрация медицинских изделий
С 01 января 2013 года вступили в силу положения Федерального закона от 21.11.2011 №323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» в отношении государственной регистрации медицинских изделий (статья 38)
Слайд 3Определение медицинских изделий
. Медицинскими изделиями являются любые инструменты, аппараты, приборы, оборудование, материалы и прочие изделия, применяемые в медицинских целях отдельно или в сочетании между")
Слайд 4Определение медицинских изделий (Федеральный закон от 21.11.11 №323-ФЗ «Об основах здоровья граждан в Российской Федерации»)
Медицинскими изделиями являются любые инструменты, аппараты, приборы, оборудование, материалы и прочие изделия, применяемые в медицинских целях отдельно или в сочетании между собой, а также вместе с другими принадлежностями, необходимыми для применения указанных изделий по назначению, включая специальное программное обеспечение, и предназначенные производителем для профилактики, диагностики, лечения и медицинской реабилитации заболеваний, мониторинга состояния организма человека, проведения медицинских исследований, восстановления, замещения, изменения анатомической структуры или физиологических функций организма, предотвращения или прерывания беременности, функциональное назначение которых не реализуется путем фармакологического, иммунологического, генетического или метаболического воздействия на организм человека. Медицинские изделия могут признаваться взаимозаменяемыми, если они сравнимы по функциональному назначению, качественным и техническим характеристикам и способны заменить друг друга.
Медицинские изделия подразделяются на классы в зависимости от потенциального риска их применения и на виды в соответствии с номенклатурной классификацией медицинских изделий. Номенклатурная классификация медицинских изделий утверждается уполномоченным федеральным органом исполнительной власти.

Слайд 5Постановления Правительства РФ, регламентирующие отношения в сфере регистрации МИ
Слайд 6Приказы МЗ РФ, регламентирующие отношения в регистрации МИ
Слайд 7Приказы Росздравнадзора, Информационные письма и Методические рекомендации
Слайд 8РУ с установленным сроком действия, выданные до 01.01.2013, действуют до истечения указанного в них срока действия. РУ бессрочного действия, выданные до 01.01.2013, действительны и подлежат замене до 01.01.2017 на РУ по форме, утверждаемой Росздравнадзором. Замена РУ осуществляется без прохождения процедуры государственной регистрации МИ на основании заявления, представленного заявителем в Росздравнадзор, с указанием сведений, предусмотренных ПП РФ. Государственная регистрация МИ, представленных на государственную регистрацию до дня вступления в силу ПП РФ, осуществляется на основании документов, представленных до дня вступления в силу ПП РФ, а также заявления о государственной регистрации МИ, представленного заявителем в соответствии с ПП РФ.
Регистрация МИ Переходный период (ПП РФ от 27.12.2012 №1416)

Слайд 9С 01.07.2012 Росздравнадзор вносит в Государственный реестр сведения, содержащиеся в комплектах регистрационной документации на вновь зарегистрированные МИ: наименование МИ регистрационный номер и дата класс потенциального риска код Общероссийского классификатора продукции наименование и место нахождения организации-заявителя наименование и место нахождения организации-изготовителя
Ведение Государственного реестра МИ и организаций, осуществляющих производство и изготовление МИ (ПП РФ от 19.06.2012 №615)

Слайд 10В настоящее время Перечень видов медицинских изделий находится в стадии разработки. Согласно письму Департамента лекарственного обеспечения и регулирования обращения медицинских изделий Минздрава России от 13.03.2013 № 277/25-3 в период до завершения разработки Перечня, медицинские изделия следует классифицировать по цифровым кодам ААА, ББ, ВВ, ГГ, установленным в приказе Минздрава России от 06.06.2012 № 4н.
Номенклатурная классификация медицинских изделий, утвержденная приказом Минздрава России от 06.06.2012 № 4н
Слайд 11технические испытания токсикологические исследования Клинические испытания испытания в целях утверждения типа средств измерений (в отношении МИ, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений, перечень которых утверждается Минздравом России).
Испытания в целях регистрации МИ
. ЗАЯВИТЕЛЬ (документы для государственной регистрации МИ). Проверка комплектности (3 дня). Уведомление об устранении нарушений (30 дней). В случае не устранения нарушений – ВОЗВРАТ ЗАЯВЛЕНИЯ И ДОКУМ")
Слайд 12СХЕМА РЕГИСТРАЦИИ МИ СОГЛАСНО ПП РФ от 27.12.2012 № 1416 редакции ПП РФ № 670 от 17.07.2014 ( I этап)
ЗАЯВИТЕЛЬ (документы для государственной регистрации МИ)
Проверка комплектности (3 дня)
Уведомление об устранении нарушений (30 дней)
В случае не устранения нарушений – ВОЗВРАТ ЗАЯВЛЕНИЯ И ДОКУМЕНТОВ
РЗН
Принятие решения о начале регистрации МИ (3 дня)
Задание на проведение экспертизы
Проведение экспертизы представленных документов организациями, подведомственными Росздравнадзору (I этап, 20 раб. дней) (определение возможности (невозможности) проведения клинических испытаний)
Заключение экспертной организации
Положительное заключение – направление на проведение клинических испытаний
Отрицательное заключение – отказ в государственной регистрации МИ
ЭУ
ЗАПРОС, содержащий сведения с указанием характера замечаний и способа их устранения
Направление ЗАПРОСА заявителю (2 дня)
ОТВЕТ на запрос (50 дней)
В случае не предоставления ОТВЕТА на запрос – ОТРИЦА- ТЕЛЬНОЕ ЗАКЛЮЧЕНИЕ
Направление ОТВЕТА в ЭУ (2 дня)
, заключение об этической обоснованности проведения КИ, (Совет по этике Минздрава России) Клинические испытания проводятся в виде: испытаний с участием человека (пациента) оценка и анализ клинических данных О начале кли")
Слайд 13Клинические испытания МИ
Основания: разрешение на проведение КИ (Росздравнадзор), заключение об этической обоснованности проведения КИ, (Совет по этике Минздрава России) Клинические испытания проводятся в виде: испытаний с участием человека (пациента) оценка и анализ клинических данных О начале клинических испытаний МИ заявитель уведомляет Росздравнадзор в течении 5 рабочих дней с начала их проведения
Слайд 14клинические испытания МИ проводятся в медицинских организациях, отвечающих требованиям, утвержденным Минздравом России установление соответствия медицинских организаций требованиям осуществляется Росздравнадзором перечень медицинских организаций, имеющих право проводить клинические испытания МИ, и реестр выданных разрешений на проведение клинических испытаний размещается на официальном сайте Росздравнадзора
. Возобновление государственной регистрации (2 дня). Проведение экспертизы представленных документов организациями, подведомственными Росздравнадзору (I Iэтап, 10 раб. дней) (экспертиза полноты и результатов проведенных испытаний и и")
Слайд 15СХЕМА РЕГИСТРАЦИИ МИ СОГЛАСНО ПП РФ от 27.12.2012 № 1416 ( II этап)
Возобновление государственной регистрации (2 дня)
Проведение экспертизы представленных документов организациями, подведомственными Росздравнадзору (I Iэтап, 10 раб. дней) (экспертиза полноты и результатов проведенных испытаний и исследований)
Положительное заключение – принятие решения о государственной регистрации (10 дней)
Отрицательное заключение – отказ в государственной регистрации МИ (10 дней)
ЭКСПЕРТНОЕ УЧРЕЖДЕНИЕ
Внесение информации в государственный реестр МИ (1 день)
ЗАЯВИТЕЛЬ (результаты клинических испытаний)

Слайд 16изделий
Нормативные правовые акты, определяющие порядок и размер оплаты экспертизы качества, эффективности и безопасности МИ в целях их государственной регистрации
Приказ МЗ РФ от 22.11.2011 №1386н «Об утверждении методики определения размера платы за экспертизу и испытания медицинских изделий в целях государственной регистрации медицинских изделий и предельных размеров платы за экспертизу и испытания медицинских изделий в целях государственной регистрации медицинских изделий»
Размеры государственной пошлины за экспертизу качества, эффективности и безопасности медицинских изделий в целях государственной регистрации медицинских изделий

Слайд 17Классификация МИ в зависимости от степени потенциального риска применения
ПРИКАЗ МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ от 06.06.2012 № 4н "Об утверждении номенклатурной классификации медицинских изделий"
Класс 3 – медицинские изделия с высокой степенью риска. Класс 2б – медицинские изделия с повышенной степенью риска. Класс 2а – медицинские изделия со средней степенью риска. Класс 1 – медицинские изделия с низкой степенью риска.
Отдельно установлены правила классификации медицинских изделий для диагностики in vitro, соответствующие рекомендациям Группы глобальной гармонизации регулирования медицинских изделий (GHTF/SG1/N045:2008).
 представляет либо направляет в регистрирующий орган заявление о государственной регистрации медицинск")
Слайд 18Порядок оформления документов
Для государственной регистрации медицинского изделия разработчик, производитель медицинского изделия или уполномоченный представитель производителя (далее - заявитель) представляет либо направляет в регистрирующий орган заявление о государственной регистрации медицинского изделия, а также документы, указанные в пункте 10 Правил: а) копия документа, подтверждающего полномочия уполномоченного представителя производителя; б) сведения о нормативной документации на медицинское изделие; в) техническая документация производителя на медицинское изделие; (в ред. Постановления Правительства РФ от 17.10.2013 N 930) г) эксплуатационная документация производителя на медицинское изделие, в том числе инструкция по применению или руководство по эксплуатации медицинского изделия; (в ред. Постановления Правительства РФ от 17.10.2013 N 930) д) фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 x 24 сантиметра); (в ред. Постановления Правительства РФ от 17.10.2013 N 930) е) документы, подтверждающие результаты технических испытаний медицинского изделия; ж) документы, подтверждающие результаты токсикологических исследований медицинского изделия, использование которого предполагает наличие контакта с организмом человека; з) документы, подтверждающие результаты испытаний медицинского изделия в целях утверждения типа средств измерений (в отношении медицинских изделий, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений, перечень которых утверждается Министерством здравоохранения Российской Федерации); и) опись документов. к) сведения, подтверждающие клиническую эффективность и безопасность медицинских изделий (в случае, если имеются); (в ред. Постановления Правительства РФ от 17.07.2014 № 670) л) проект плана клинических испытаний медицинского изделия с обосновывающими его материалами (в случае, если имеется) (в ред. Постановления Правительства РФ от 17.07.2014 № 670)
 наименование медицинского изделия (с указанием принадлежностей, необходимых для применения медицинского изделия по назначению); б) в отношении разработчика - полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, ор")
Слайд 19Заявление о государственной регистрации
а) наименование медицинского изделия (с указанием принадлежностей, необходимых для применения медицинского изделия по назначению); б) в отношении разработчика - полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес (место нахождения), а также номера телефонов и (в случае, если имеется) адрес электронной почты юридического лица; в) в отношении производителя медицинского изделия - полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес (место нахождения), а также номера телефонов и (в случае, если имеется) адрес электронной почты юридического лица; г) в отношении уполномоченного представителя производителя - полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес (место нахождения), а также номера телефонов и (в случае, если имеется) адрес электронной почты юридического лица; д) в отношении юридического лица, на имя которого может быть выдано регистрационное удостоверение, - полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес (место нахождения), а также номера телефонов и (в случае, если имеется) адрес электронной почты юридического лица; е) место производства медицинского изделия; ж) назначение медицинского изделия, установленное производителем; з) вид медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий; и) класс потенциального риска применения медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий; к) код Общероссийского классификатора продукции для медицинского изделия; л) сведения о способе получения регистрационного удостоверения, а также информации, связанной с процедурой государственной регистрации медицинского изделия.

Слайд 20Техническая документация
Документ или документы, содержащие следующую информацию: - сведения регламентирующие конструкцию МИ - технические требования к МИ - данные для разработки МИ - данные для производства МИ - данные для применения МИ - данные для эксплуатации МИ - данные для технического обслуживания МИ - данные для ремонта МИ - данные для утилизации или уничтожения МИ
Слайд 21Примеры технической документации
- Технические условия (РФ, Беларусь, Украина) - Технический файл - Выписка из технического файла Документ под наименованием «Техническая документация» - другое
Слайд 22Эксплуатационная документация
Документ или документы, содержащие следующую информацию: Информацию для ознакомления потребителя с конструкцией медицинского изделия Условия и правила эксплуатации: - использование по назначению - техническое обслуживание - текущий ремонт - хранение - транспортировка
Слайд 23Документ или документы, содержащие следующую информацию: - гарантированные производителем значения основных параметров, характеристик (свойств) - гарантийные обязательства - сведения о его утилизации или уничтожении Примеры эксплуатационной документации: - Руководство/Инструкция по эксплуатации - Руководство/Инструкция пользователя - Руководство/Инструкция по применению - Этикетка Другое Техническая и эксплуатационная документация представляется производителем (ПП РФ №930)!!!

Слайд 24В целях оперативного информирования по вопросам направления уведомлений о ходе рассмотрения документов и принятых решений о регистрации медицинских изделий/внесении изменений, Росздравнадзором создан дополнительный электронный сервис в «кабинете заявителя», размещенный на сайте Росздравнадзора по адресу http://www.roszdravnadzor.ru/national_foreign_medprod/Reg_med_products/cabinet_for_applicant(медицинские изделия (изделия медицинского назначения)/регистрация медицинских изделий (изделий медицинского назначения)/кабинет заявителя). Сервис позволяет получать копии отсканированных документов о принятых решениях в ходе регистрации.
 не допускается истребование экспертным учреждением у заявителя либо иных лиц материало")
Слайд 25п. 21 Постановления № 1416 в редакции Постановления № 670 от 17.07.2014
Дополнен пункт следующим содержанием: При проведении экспертизы качества, эффективности и безопасности медицинского изделия (на любом этапе) не допускается истребование экспертным учреждением у заявителя либо иных лиц материалов, необходимых для проведения экспертизы при недостаточности предоставленных заявителем материалов и сведений в соответствии с п. 10 Постановления № 1416, эксперт ставит вопрос о предоставлении ему необходимых материалов и сведений перед руководителем экспертного учреждения. руководитель экспертного учреждения обращается с соответствующим ЗАПРОСОМ в регистрирующий орган, выдавши задание на проведение экспертизы (Росздравнадзор). регистрирующий орган в течение 2 РАБОЧИХ ДНЕЙ со дня поступления ЗАПРОСА направляет его заявителю. Примечания: 1) ЗАПРОС содержит необходимые сведения с указанием характера замечаний и способа их устранения. 2) ЗАПРОС направляется однократно и может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи либо в форме электронного документа, подписанного электронной подписью. заявитель обязан предоставить представить ОТВЕТ на ЗАПРОС регистрирующего органа в срок, не превышающий 50 РАБОЧИХ ДНЕЙ со дня получения этого ЗАПРОСА. регистрирующий орган направляет ОТВЕТ заявителя в экспертное учреждение в течение 2 РАБОЧИХ ДНЕЙ со дня поступления от заявителя ответа на запрос. Примечание: В случае непредставления по истечении 50 рабочих дней заявителем ответа на запрос регистрирующий орган в течение 2 рабочих дней направляет в экспертное учреждение уведомление о непредставлении заявителем ответа на запрос регистрирующего органа для подготовки заключения экспертного учреждения на основании документов, имеющихся в его распоряжении. Время со дня направления запроса регистрирующего органа до дня получения ответа на запрос или уведомления о непредставлении ответа на запрос не учитывается при исчислении срока проведения экспертизы качества, эффективности и безопасности медицинского изделия.

Слайд 26п. 38 Постановления № 1416 в редакции Постановления № 670 от 17.07.2014
Дополнен пункт следующим содержанием: Для внесения изменений в регистрационное удостоверение заявитель не позднее чем через 30 рабочих дней со дня внесения соответствующих изменений представляет либо направляет в регистрирующий орган заявление о внесении изменений в регистрационное удостоверение (далее - заявление о внесении изменений), оформленное в соответствии с пунктом 9 настоящих Правил, с приложением указанных изменений и подтверждения, что внесение изменений в регистрационное удостоверение не влечет изменения свойств и характеристик, влияющих на качество, эффективность и безопасность медицинского изделия, или совершенствует свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия, и следующие документы: а) копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя); б) номер регистрационного досье; в) опись документов.
 внесения изменений в документы, предусмотренные подпунктом \"а\" пункта 54 настоящих Правил, заявитель направляет в регистрирующий орга")
Слайд 27п. 55 Постановления № 1416 в редакции Постановления № 670 от 17.07.2014
Дополнен пункт следующим содержанием: В случае необходимости (по желанию заявителя) внесения изменений в документы, предусмотренные подпунктом "а" пункта 54 настоящих Правил, заявитель направляет в регистрирующий орган заявление о внесении изменений с представлением документов, подтверждающих такие изменения. В случае необходимости внесения изменений в документы, указанные в подпунктах "в" и "г" пункта 10 настоящих Правил, внесение изменений проводится по результатам экспертизы, проведенной в порядке, аналогичном порядку проведения экспертизы качества, эффективности и безопасности медицинского изделия для его государственной регистрации в соответствии с пунктом 21 настоящих Правил. 55(1). Основаниями для вынесения экспертным учреждением заключения о невозможности внесения изменений в документы, предусмотренные подпунктами "в" и "г" пункта 10 настоящих Правил, являются: а) недостоверность представленных сведений, обосновывающих внесение изменений; б) отсутствие сведений, подтверждающих неизменность функционального назначения и (или) принципа действия медицинского изделия, в связи с вносимыми изменениями в документацию. 55(2). Регистрирующий орган в течение 2 рабочих дней со дня получения экспертного заключения принимает решение о возможности (невозможности) внесения изменений в документы, предусмотренные подпунктом "а" пункта 54 настоящих Правил, и уведомляет о своем решении заявителя по почте заказным письмом или в электронной форме по телекоммуникационным каналам связи. Основанием для принятия решения об отказе во внесении изменений в документы на медицинское изделие является получение регистрирующим органом от экспертного учреждения заключения о невозможности внесения изменений в документы на медицинское изделие. Хранение регистрационного досье осуществляется регистрирующим органом в порядке, установленном законодательством Российской Федерации об архивном деле.

Слайд 28СПАСИБО ЗА ВНИМАНИЕ!
 Слайд 1
Слайд 1 Слайд 2
Слайд 2 Слайд 3
Слайд 3 Слайд 4
Слайд 4 Слайд 5
Слайд 5 Слайд 6
Слайд 6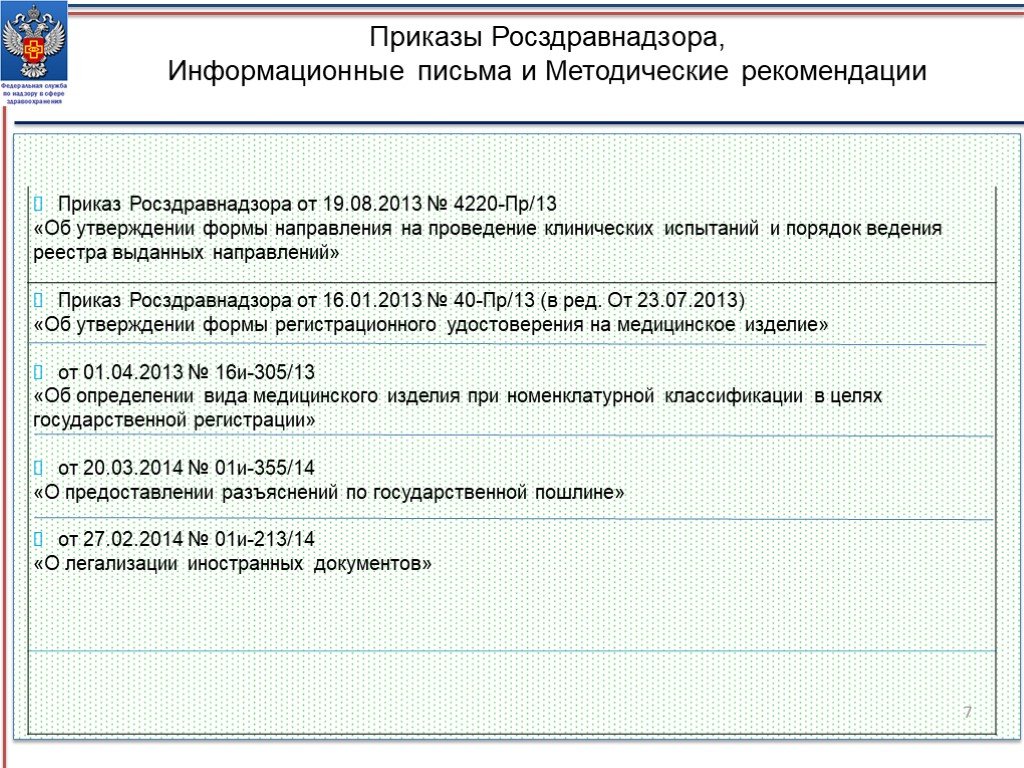 Слайд 7
Слайд 7 Слайд 8
Слайд 8 Слайд 9
Слайд 9 Слайд 10
Слайд 10 Слайд 11
Слайд 11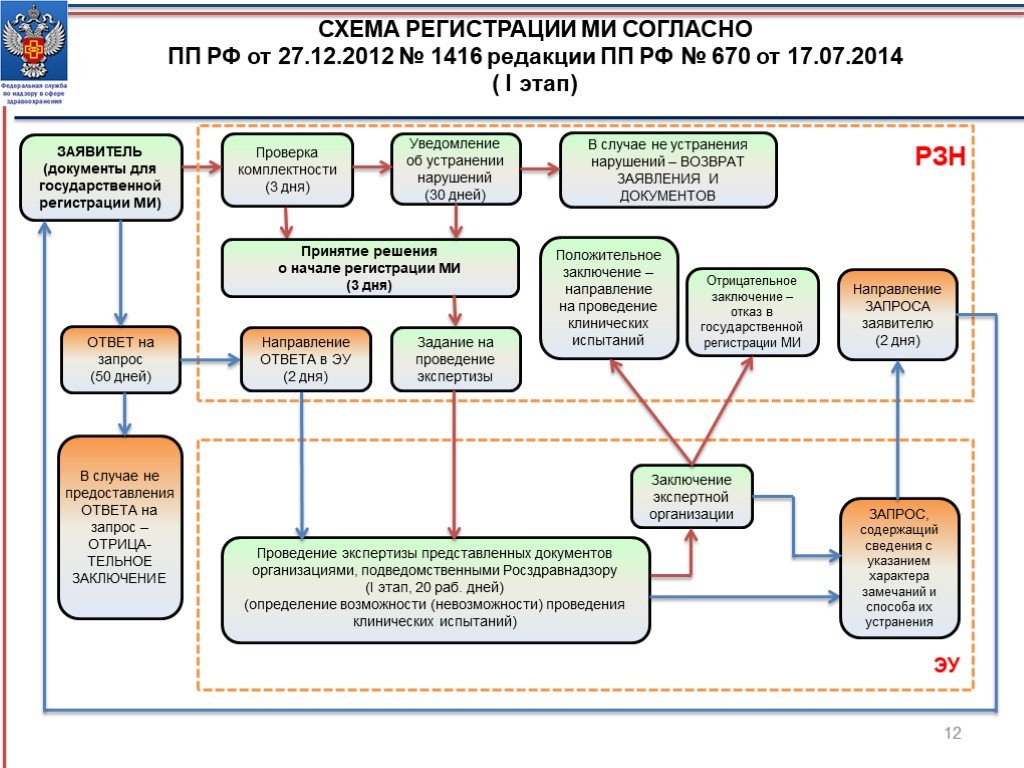 Слайд 12
Слайд 12 Слайд 13
Слайд 13 Слайд 14
Слайд 14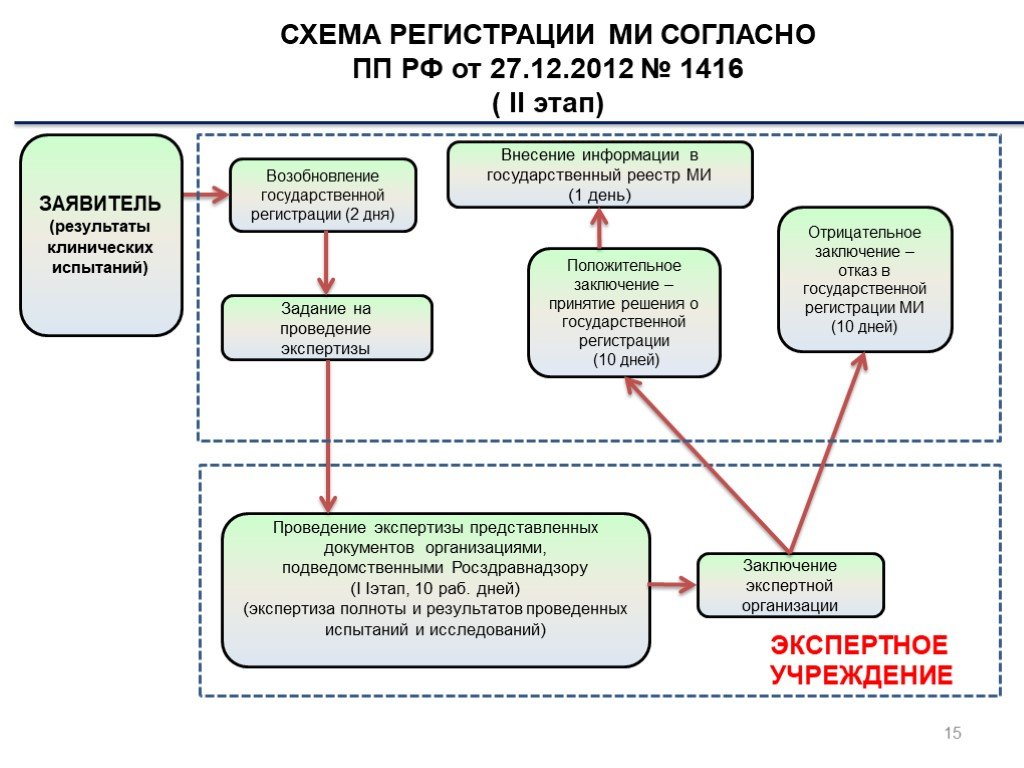 Слайд 15
Слайд 15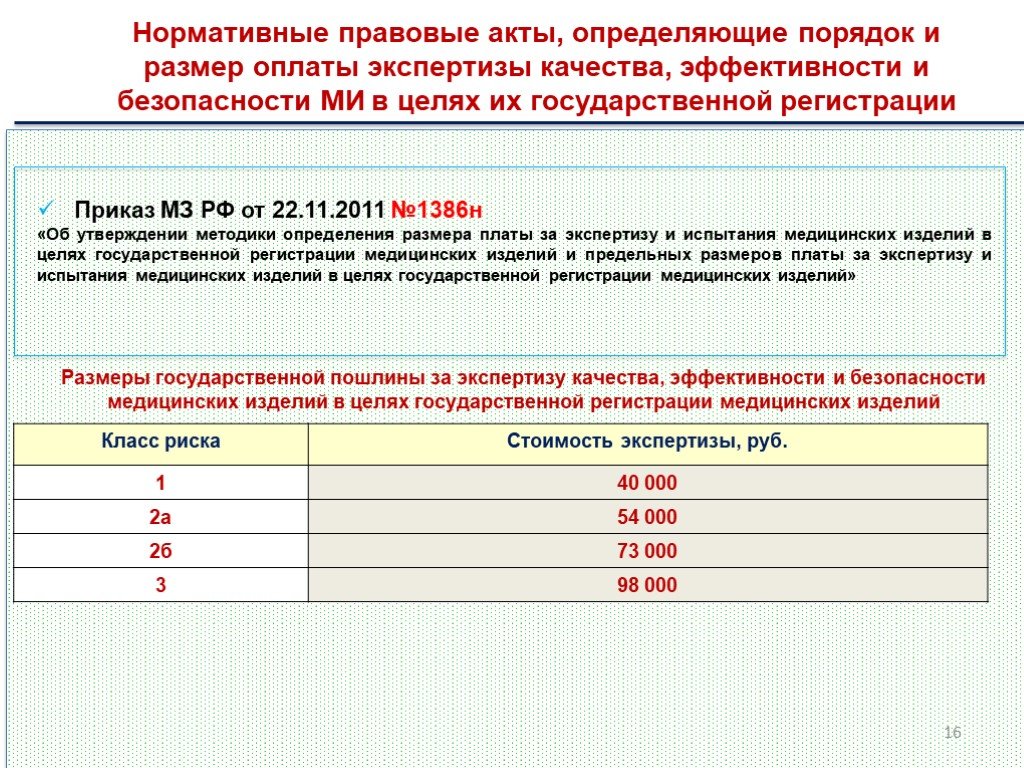 Слайд 16
Слайд 16 Слайд 17
Слайд 17 Слайд 18
Слайд 18 Слайд 19
Слайд 19 Слайд 20
Слайд 20 Слайд 21
Слайд 21 Слайд 22
Слайд 22 Слайд 23
Слайд 23 Слайд 24
Слайд 24 Слайд 25
Слайд 25 Слайд 26
Слайд 26 Слайд 27
Слайд 27 Слайд 28
Слайд 28




























