Слайд 1Наследственные пигментные гепатозы
Выполнила: Шамрай В.Ю. 6/114 ВБ Проверила: Нагашбаева М.К. Астана - 2015
АО «Медицинский Университет Астана»
Слайд 2Пигментные гепатозы (доброкачественные гипербилирубинемии) — заболевания, связанные с наследственным нарушением обмена билирубина, проявляющиеся хронической или перемежающейся желтухой без выраженного изменения структуры и функции печени и явных признаков повышенного гемолиза и холестаза.
Слайд 3МОРФОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА
сохраняет гистологическое строение, близкое к норме, каких-либо признаков диспротеиноза, некроза печеночных клеток, как правило, нет. не выявлено грубых морфологических изменений Патогномонично - накопление липофусцина
Слайд 4Накопление липофусцина
накопление в печеночных клетках по ходу желчных капилляров мелкого золотистого и желтовато-коричневого пигмента.
Слайд 5Пигмент концентрируется преимущественно в центре долек, а в случае особенно большого накопления - в промежуточных и периферических отделах долек. При гипербилирубинемии типа синдрома Жильбера - накопление пылевидного золотистого коричневого пигмента . При синдроме Дабина—Джонсона - накопление в центре долек более грубых зерен темно-коричневого пигмента.
Слайд 6Гистохимические свойства, характерные для хромолипоидов — липофусцинов:
Пигмент окрашивается черным суданом в парафиновых срезах, что свидетельствует о присутствии в нем фосфолипидов. положительный тест Шморля - восстанавливает феррицианид в феррацианид. Гранулами пигмента красноватого оттенка может указывать на присутствие высокомолекулярных ненасыщенных жирных кислот. ШИК-реакция и реакция тетразониевого сочетания отрицательны. Активность окислительных ферментов в центре долек, где происходит максимальное накопление пигмента, понижается.

Слайд 7В стенках синусоидов установлена умеренная реакция на щелочную фосфатазу. Активность фермента заметна только в синусоидах, окружающих центральные вены, а также расположенных вблизи портальных трактов. В печеночных клетках активность фермента отсутствует. При реакции на кислую фосфатазу активность фермента оказалась более высокой в центре долек, чем на периферии, там, где концентрируются гранулы липофусцина. Накопление липофусцина нередко сочетается со своеобразной мелкокапельной (мелкозернистой) жировой дистрофией. Эти капельки жира, образующиеся при распаде клеточных ультраструктур (митохондрий), в дальнейшем превращаются в зерна липофусцина. Таким образом, пылевидное ожирение можно рассматривать как стадию образования липофусцина. В пунктатах печени с наибольшим отложением пигмента жировая дистрофия или отсутствует, или бывает очаговой.

Слайд 8При электронно-микроскопическом исследовании
Разрежение цитоплазмы; она выглядит оптически прозрачной с большим количеством крупных вакуолей диаметром до 1,5—2 мкм. Эндоплазматическая сеть представлена везикулярными фрагментами, среди которых можно различить отдельные структуры, образованные зернистыми мембранами. Митохондрии плотные, кристы выделяются нечетко. Отложение плотного глыбчатого материала, по-видимому, кальциевых солей.
Слайд 9КЛИНИЧЕСКАЯ КАРТИНА
имеют семейный характер и обусловлены недостаточностью ферментов, ответственных за захват, конъюгацию или экскрецию билирубина Гипербилирубинемия вызвана преимущественно нарушением одной из фаз внутрипеченочного обмена билирубина.
Слайд 10Основными заболеваниями являются
Синдромы Жильбера Синдром Криглера—Найяра Синдром Дабина—Джонсона Синдром Ротора
Слайд 11СИНДРОМ ЖИЛЬБЕРА
Заболевание, которое обусловлено мутацией гена, кодирующего фермент уридиндифосфат. Генетически обусловленного понижения захвата и конъюгации билирубина. Выявляется в детском и юношеском возрасте до 25 лет Мужчины и женщины – 10:1 Врачи, студенты-медики, инженеры.
Слайд 12Синдром Жильбера характеризуется следующими факторами:
Частичная недостаточность глюкуронилтрансферазы (активность порядка тридцати процентов от нормы) Умеренное повышение содержания билирубина в крови Уменьшение повышенного билирубина под воздействием фенобарбитала Отсутствие других морфологических или функциональных изменений печени Аутосомно – доминантный тип наследования
Слайд 13Желтуха - иктеричность склер. Характерна матово-желтушная кожа, особенно лица. Характерна триада: печеночная маска, ксантелазмы век и желтый цвет кожи. Боль или чувство тяжести в правом подреберье наблюдаются часто, особенно в периоды обострений. Диспепсические явления Астеновегетативные расстройства: депрессия, неспособность концентрировать внимание, быстрая утомляемость, слабость, головокружение, потливость, плохой сон, неприятные ощущения в области сердца — наблюдаются почти постоянно.
Слайд 15Увеличение печени - выступает на 1—2 см из-под правого подреберья по срединно-ключичной линии, в отдельных случаях —на 3—4 см, консистенция ее мягкая, пальпация безболезненная. Инфекция в желчных путях Увеличение гемоглобина свыше 160 г/л и числа эритроцитов выявлено больных гипербилирубинемией.
Слайд 17Синдром Криглера - Найяра
Синдром Криглера - Найяра - врождённaя наследственная злокачественная неконъюгированная гипербилирубинемия, характеризующaяся желтухой и тяжёлым поражением нервной системы. С равной частотой встречается у мальчиков и девочек. Тип наследования аутосомно-рецессивный
Слайд 18Этиология
Относится к разряду печеночных паренхиматозных желтух. Характер течения зависит от типа (разновидности) синдрома. В основе заболевания лежит генетический дефект, наследуемый по аутосомно-рецессивному типу и заключающийся в частичном или полном отсутствии глюкуронилтрансферазы.
Слайд 19Патогенез
Гипербилирубинемия является следствием нарушения конъюгации в печени билирубина с глюкуроновой кислотой, обусловленного отсутствием или значительной недостаточностью фермента глюкуронилтрансферазы.
Слайд 20Выделяют два варианта синдрома:
1-й тип: полное отсутствие активности глюкуронилтрансферазы. Обусловлен мутациями в кодирующей последовательности гена UGTIAI, что приводит к образованию неполноценного фермента уридиндифосфатглюкуронидазы, который разрушается. В связи с чем реакции глюкунизации билирубина не происходит и непрямой билирубин накапливается в организме, в том числе ядрах серого вещества, обуславливая тяжёлую клинику заболевания.
Слайд 222-й тип: синдром Ариаса - активность фермента менее 20 % от нормальной. Заболевание также обусловлено мутациями в кодирующей последовательности гена UGTIAI. Больные часто являются компаундгетерозиготами, имеющими в одной хромосоме инсерцию в промотере, а в другой миссенс-мутацию в экзоне. Кроме того описаны пациенты несущие инсерции в промоторной области гена UGTIAI в гомозиготном состоянии в сочетании со структурной мутацией в экзоне.

Слайд 23Симптомы Синдрома Криглера - Найяра
I тип: характеризуется злокачественный прогрессирующим течением. Манифестация наступает в первые часы жизни. Клинические проявления: желтушность склер и кожных покровов, судороги, опистотонус, нистагм, атетоз, замедление умственного развития (билирубиновая энцефалопатия), на ЭЭГ регистрируется медленная активность в задних долях и параксизмальная активность. Биохимические показатели: уровень билирубина в крови выше 200 мкмоль/л. В желчи полностью отсутствует конъюгированный билирубин. В отсутствии лечебных мероприятий больные погибают в течение первого года жизни от ядерной желтухи.
, ребенок начинает плохо сосать, лежит в расслабленной позе, резко реагирует на слабые раздражители, дыхание становится редким с длительными периода")
Слайд 241 фаза билирубиновой энцефалопатии. В первой фазе энцефалопатии наблюдается угнетение безусловно-рефлекторной деятельности (апатия, вялость, сонливость), ребенок начинает плохо сосать, лежит в расслабленной позе, резко реагирует на слабые раздражители, дыхание становится редким с длительными периодами остановки. Могут отмечаться: монотонный крик, срыгивания, рвота, «блуждающий взгляд». Наблюдается цианоз.

Слайд 252 фаза билирубиновой энцефалопатии. Вторая фаза продолжается от нескольких дней до нескольких недель. В этой фазе развивается клиническая картина поражения ядер головного мозга. Наблюдаются спастичность, ригидность затылочных мышц, вынужденное положение тела с опистотонусом. Характерно вынужденное положение тела с «негнущимися» конечностями и сжатыми в кулаки кистями. Ребенок пронзительно кричит, у него отмечается выбухание большого родничка, подергивание мышц лица, крупноразмашистый тремор рук, исчезновение видимой реакции на звук, сосательного рефлекса. Наблюдаются нистагм, апноэ, брадикардия, летаргия, судороги.
 формируются стойкие неврологические нарушения: параличи, парезы, нистагм, атетоз. Наблюдается")
Слайд 273 фаза билирубиновой энцефалопатии. Третья фаза период ложного благополучия. Явления спастичности полностью или частично исчезают. 4 фаза билирубиновой энцефалопатии. В четвертой фазе (на 3-5 месяце жизни) формируются стойкие неврологические нарушения: параличи, парезы, нистагм, атетоз. Наблюдается грубое отставание в физическом и психическом развитии: ребенок не держит голову, не реагирует на голос матери и другие звуковые раздражители; не следит за игрушкой. Смерть пациентов при синдроме I типа обусловлена развитием билирубиновой энцефалопатии и наступает в течение первых 2 лет жизни. В редких случаях больные с синдромом Криглера-Найяра I типа доживают до подросткового возраста.

Слайд 28II тип: занимает промежуточное положение по тяжести клинических проявлений между синдромом Криглера-Найяра I типа и синдромом Жильбера. Манифестация наступает несколько позже, чем при I типе, от нескольких месяцев до первых лет. У ряда больных желтуха может не проявляться до подросткового возраста, и неврологические осложнения наблюдаются редко; в некоторых случаях клиническая симптоматика отсутствует. Клинические проявления сходны с 1 типом, но менее тяжёлые. Редко, при интеркуррентных инфекциях или в условиях стресса у больных с синдромом Криглера-Найяра II типа может возникать билирубиновая энцефалопатия. Биохимические показатели: уровень билирубина в крови менее 200 мкмоль/л. Желчь пигментирована и содержит билирубин-глюгуронид. Проба с фенобарбиталом положительная.
Слайд 29Синдром Дабина-Джонсона
Синдром Дабина-Джонсона – хроническое наследственное заболевание, проявляющееся непостоянной желтухой. Синдром Дабина-Джонсона имеет аутосомно-рецессивный тип наследования.
Слайд 30Генетический дефект заключается в появлении мутации в гене, кодирующем белок, который является ионным каналом, транспортером органических анионов (cMOAT). В результате гепатобилиарный транспорт билирубина и органических анионов нарушается. В крови увеличивается содержание фракции конъюгированного билирубина, в моче – билирубинурия.
Слайд 31Основным звеном патогенеза является нарушение экскреции пигмента из гепатоцитов, приводящее к регургитации билирубина.
Слайд 32Клиника
Синдром Дабина - Джонсона - очень редкое заболевание, встречающееся преимущественно у мужчин молодого возраста, в некоторых случаях - с рождения. Клиническая симптоматика более ярко выражена, чем при других формах гипербилирубинемии. Отмечают повышенную утомляемость, плохой аппетит, боли в правом подреберье вплоть до колик, поносы. Желтуха непостоянной и сопровождалась нерезким кожным зудом. Диспепсические кризы чаще, самочувствие всегда плохое. В двух наблюдениях диспепсическим расстройствам предшествовал 2-3-дневный продромальный период с легкой гиперемией зева, субфебрильной температурой. У некоторых больных заболевание десятилетиями протекает бессимптомно. Печень нормальных размеров или выступает на 1-2 см из-под края реберной дуги.

Слайд 34Печень больных не может адекватно экскротировать билирубин, бромсульфалсин и контрастные препараты для холецистографии. Вследствие этого выявляют отклонение от нормы содержания билирубина, бромсульфал ей новой пробы и активности щелочной фосфатазы, а также частое отсутствие тени желчного пузыря при холецистографии. При раздельном исследовании фракции билирубина преобладает прямой билирубин. В соответствии с этим при синдроме Дабина-Джонсона наблюдается билирубинурия.
Слайд 36Синдром Ротора
Синдром Ротора (Rotor syndrome) - наследственный пигментный гепатоз – напоминает легкую форму синдрома Дaбина — Джонсона - дефект экскреции билирубина менее выражен. Отличия: отсутствие 2-го пика на кривой выделения бромсульфалеина, обнаруживается желчный пузырь при холецистографии, не происходит образования тёмного пигмента в печеночных клетках.
Слайд 38Для наследственного синдрома Ротора характерна прямая гипербилирубинемия без нарушения активности печеночных ферментов. Синдром Ротора, как правило, возникает в детском возрасте, наследуется по аутосомно-рецессивному типу. В гепатоцитах находят признаки жировой дистрофии.
Слайд 39Проявляется интермиттирующей желтухой. Заболевание выявляется с детства, нарастает билирубин, повышается содержание копропорфирина в моче, задерживается бромсульфалеин, желчный пузырь в данном случае контрастируется. У большинства болезнь протекает бессимптомно.
Слайд 40Лечение
Терапия врожденных функциональных гипербилирубинемий пока неэффективна. Больных с наследственными гепатозами следует предупредить о необходимости полностью исключить из употребления алкогольные напитки, придерживаться разумной щадящей диеты, избегать острых блюд и физического переутомления; им не следует назначать лекарства, обладающие токсическим действием на печень.
Слайд 41Учитывая наследственный характер врожденных функциональных билирубинемий, в их профилактике большое место занимают медико-генетические консультации, так как специалисты-генетики могут оценить риск возникновения этих заболеваний у потомства и не рекомендовать родителям, страдающим функциональными повышениями уровня билирубина в крови, иметь детей, если риск передачи заболевания детям достаточно высок.
 Слайд 1
Слайд 1 Слайд 2
Слайд 2 Слайд 3
Слайд 3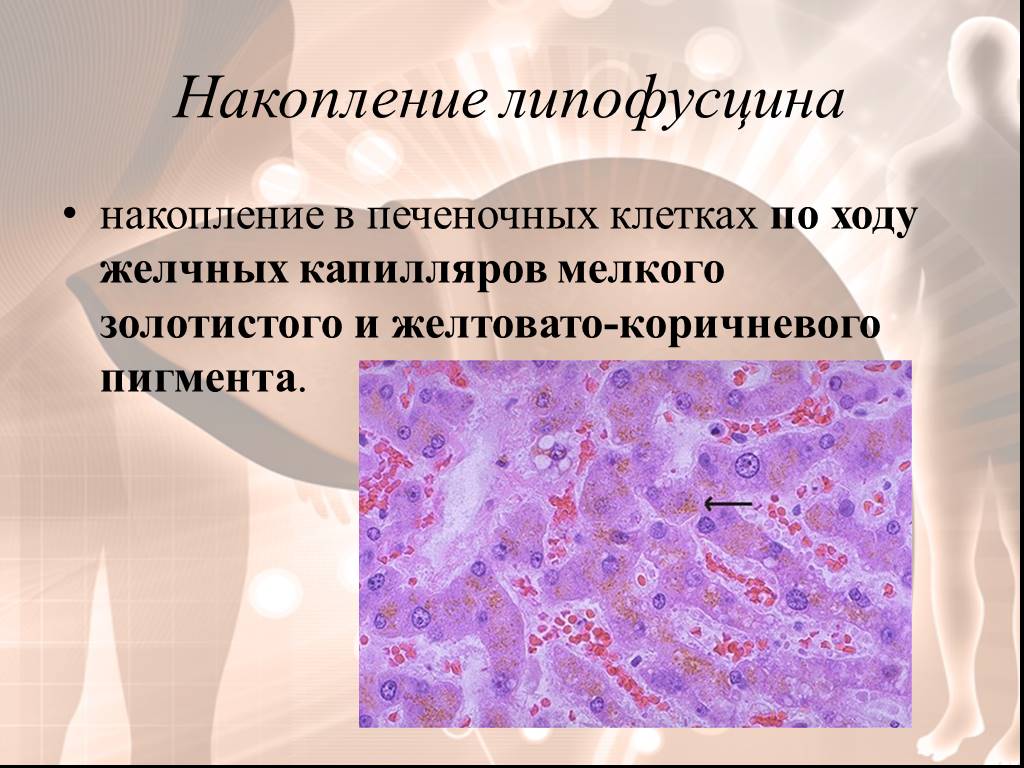 Слайд 4
Слайд 4 Слайд 5
Слайд 5 Слайд 6
Слайд 6 Слайд 7
Слайд 7 Слайд 8
Слайд 8 Слайд 9
Слайд 9 Слайд 10
Слайд 10 Слайд 11
Слайд 11 Слайд 12
Слайд 12 Слайд 13
Слайд 13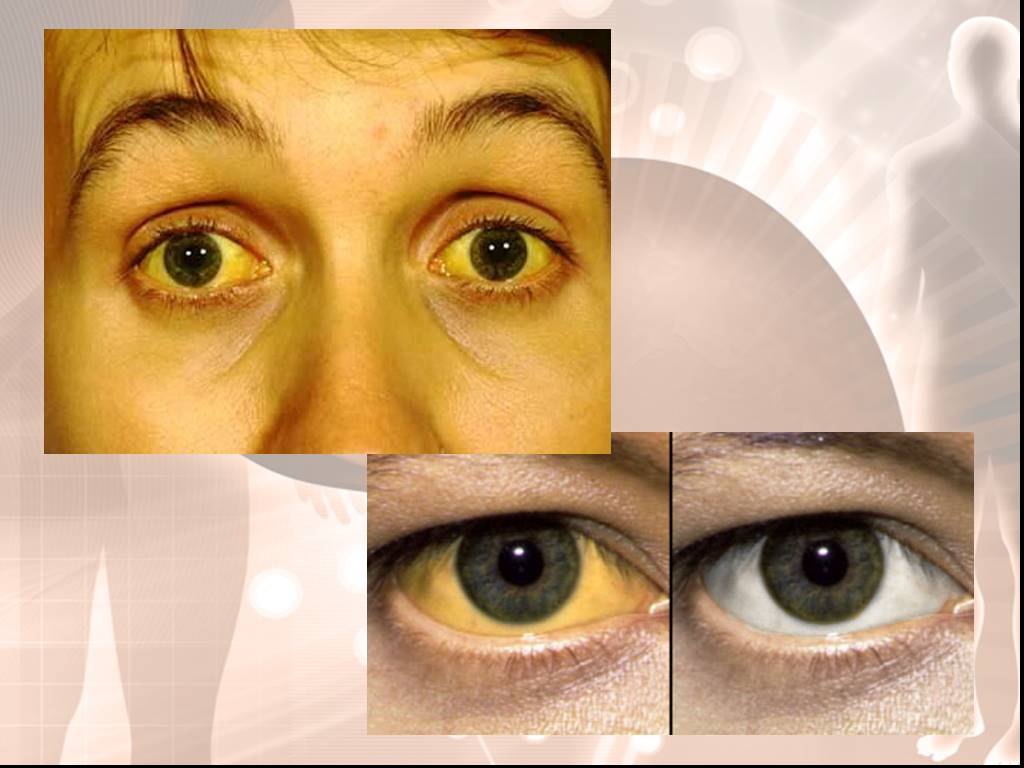 Слайд 14
Слайд 14 Слайд 15
Слайд 15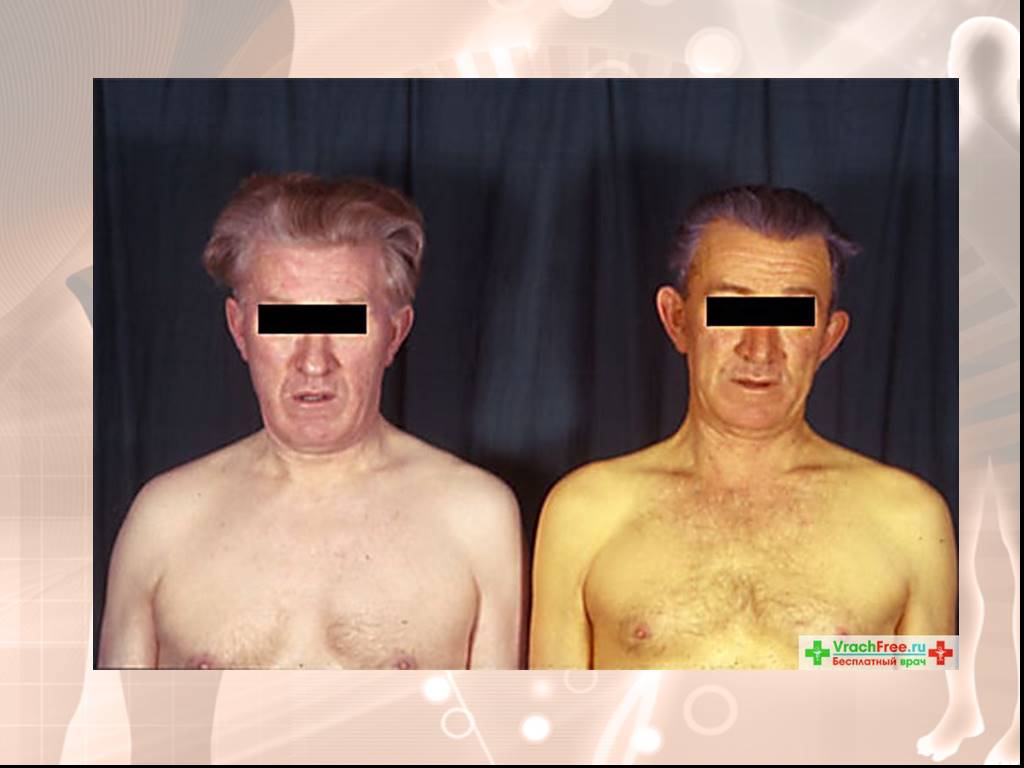 Слайд 16
Слайд 16 Слайд 17
Слайд 17 Слайд 18
Слайд 18 Слайд 19
Слайд 19 Слайд 20
Слайд 20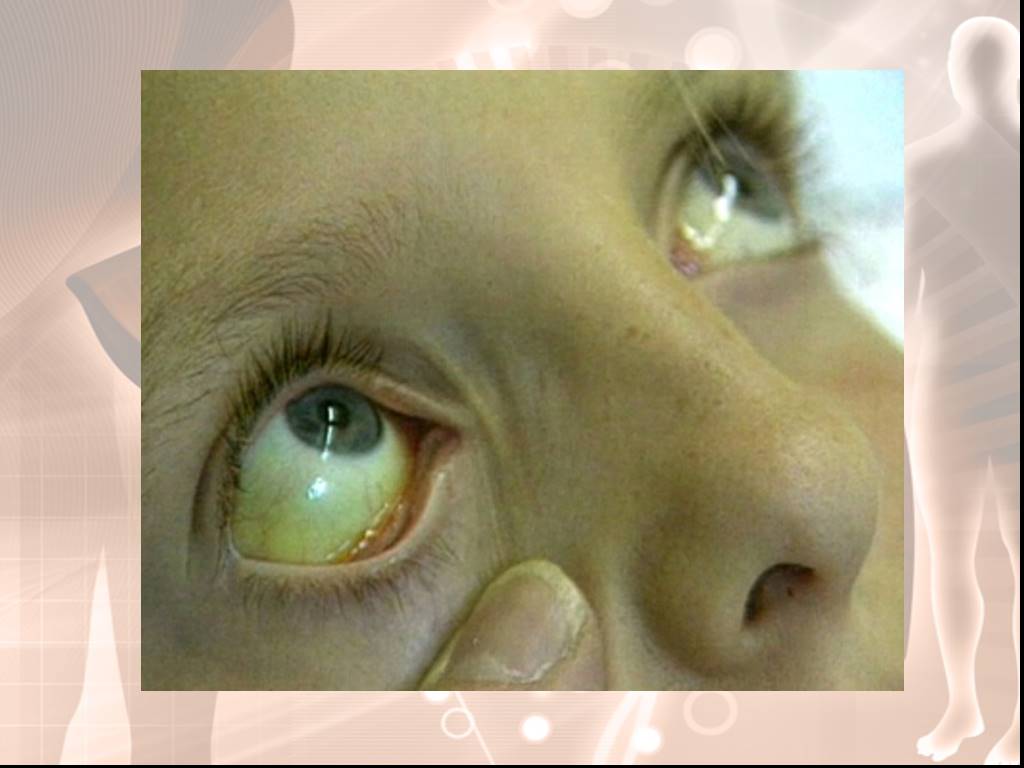 Слайд 21
Слайд 21 Слайд 22
Слайд 22 Слайд 23
Слайд 23 Слайд 24
Слайд 24 Слайд 25
Слайд 25 Слайд 26
Слайд 26 Слайд 27
Слайд 27 Слайд 28
Слайд 28 Слайд 29
Слайд 29 Слайд 30
Слайд 30 Слайд 31
Слайд 31 Слайд 32
Слайд 32 Слайд 33
Слайд 33 Слайд 34
Слайд 34 Слайд 35
Слайд 35 Слайд 36
Слайд 36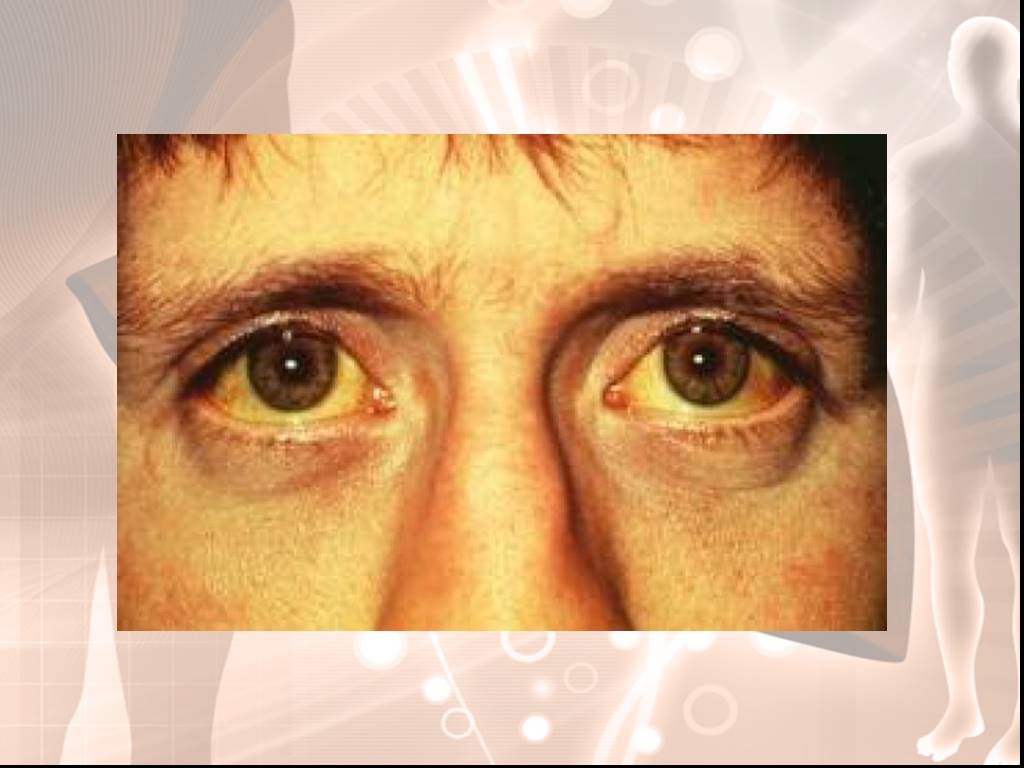 Слайд 37
Слайд 37 Слайд 38
Слайд 38 Слайд 39
Слайд 39 Слайд 40
Слайд 40 Слайд 41
Слайд 41 Слайд 42
Слайд 42











