Слайд 1Фенилкетонурия
ФКУ финилпировиноградная олигофрения болезнь Фёллинга
Слайд 2Наследственное заболевание обмена одной из важных аминокислот (фенилаланина), в связи с недостатком или полным отсутствием необходимого для обмена фермента. Это приводит к накоплению в организме особо токсичных веществ, поражающих нервную систему.
Слайд 3Местоположение гена в хромосоме (локус):
12 q 22 – 24, 2 4 р 15,3
Слайд 4Этиология и патогенез:
Слайд 5Физические признаки:
- дети белокурые со светлой кожей и голубыми глазами - часто отмечаются экзема, дерматиты - моча и пот имеют « заплесневелый», «мышиный», «волчий» запах - быстрое и чрезмерное прибавление в весе, однако остаются рыхлыми, вялыми. - у большинства рано зарастает большой родничок
Слайд 6Психопатологически отмечается:
Умственная отсталость( 65% - глубокая, 31.8% - умеренная и 3.2% - легкая) Недоразвитие речи (ее или совсем нет, или есть отдельные слова, которые больные не соотносят с объектом), т.е. нарушено: - понимание речи - звукопроизношение
Слайд 7Неврологическая симптоматика:
Эпилептиформные припадки Нарушение мышечного тонуса Плохая координация движений Много стереотипии, часты другие знаки экстрапирамидной недостаточности (атетоидные, хореиформные движения)
Слайд 8Расстройства поведения:
Двигательное беспокойство, целенаправленные, неуправляемые перемещения от объекта к объекту, бесцельные манипуляции с предметами. или Дети пассивны, вялы, плохо узнают близких, оживляются при упоминании о еде.
Слайд 9Патологоанатомически обнаруживается:
- малая масса мозга - дефекты миелинезации в коре больших полушарии( особенно в лобных и височных долях), и других структурах (внутренняя капсула, зрительные проводящие пути) - депигментация черной субстанции
Слайд 10Обследование детей:
Все новорожденные обследуются по специальным программам скрининга на повышение концентрации фенилаланина. Используют: 1.Микробиологический метод определения концентрации фенилаланина в крови. 2. Проба Фёллинга на фенилпировидноградную кислоту в моче (берется 5% раствор треххлористого железа и уксусной кислоты и прибавляется несколько капель к моче, появление зеленой окраски говорит о положительной реакции на фенилаланин). В дальнейшем определяют количественное содержание фенилаланина в крови и моче(хроматография аминокислот), , это исследование с помощью специальных анализаторов.
. Ген фенилкетонурии встречается в среднем у 1-2 на 100 человек, но болезнь может возникнуть лишь в том случае, если и мать")
Слайд 11Наследственность:
Болезнь наследуется по рецессивному типу (т.е. болеют сестры и братья из одной семьи, а родители здоровы, хотя и являются гетерозиготными носителями гена ФКУ). Ген фенилкетонурии встречается в среднем у 1-2 на 100 человек, но болезнь может возникнуть лишь в том случае, если и мать и отец ребенка являются носителями этого гена, и ребенок унаследует его в двойном наборе. Поэтому болезнь встречается значительно реже, чем распространен ген. Больные ФКУ (обладатели двух патологических генов) могут иметь детей с фенилкетонурией только при вступлении в брак с носителями таких же генов. При вступлении в брак с лицами свободными от гена ФКУ, дети не болеют этим заболеванием.
Слайд 12Диетотерапия:
Исключить: мясо, колбасы, рыбу, бульоны, яйца, творог, сыр, мучные изделия, каши из естественных круп, фасоль, орехи, шоколад. Меню для детей составляется из: фруктов, овощей, крахмальных изделий, жиров, со строгим учетом содержания в них фенилаланина.
Слайд 13MD мил ФКУ-0»
Лечебное питание на основе аминокислот без фениланина, предназначенное для вскармливания детей, больных фенилкетонурией первого года жизни.
Слайд 14«MD мил ФКУ-1»
Лечебное питание на основе аминокислот без фенилалина, предназначенное для питания больных фенилкетонурией детей в возрасте от 1 года.
Слайд 16«MD мил ФКУ-3»
Лечебное питание на основе аминокислот без фенилалина, предназначенное для питания больных фенилкетонурией - детей старше 1 года, взрослых и беременных женщин.
Слайд 17При несоблюдении женщиной специфической диеты во время беременности у ребёнка возникают следующие пороки развития:
в 92% случаев - умственная отсталость в 73% случаев - микроцефалия в 12% случаев - врождённые пороки сердца в 40% случаев - низкая масса тела при рождении
Слайд 18Профилактика фенилкетонурии:
1. Большое значение имеет специальное наблюдение за «семьями риска». Новорожденные из этих семей должны быть подвергнуты обязательному биохимическому исследованию и при показаниях к раннему лечению. 2. Внедрение программ массового скрининга новорожденных для раннего выявления ФКУ и своевременного назначения диетотерапии. 3. Пренатальная диагностика в «семьях высокого риска».
Слайд 19Работу выполнили:
Панкратова В.С. Сухарева М.В. Сионская Н.В. Зубанова Н.А. Богачева А.Н. Чилипалова И.С Киселева М.Е. Святохо И.А. Белькова Я.В. Петрова Н.Л.
Слайд 20Используемая литература:
Л.О.Бадалян «Невропатология» М. – 2003. «Краткая медицинская энциклопедия» Гл. редактор Б.В. Петровский М. – 1990. Ф.А. Самсонов «Основы генетики в дефектологии» М. – 1980. А.Ю. Асанов, Н.С.Демикова, С.А. Морозов «Основы генетики и наследственные нарушения у детей» М . – 2003 Е.М. Мастюкова, А.Г. Московкина «Основы генетики» м. – 2003 http://mame66.ru/zdorovya_baby/-fenilketonuriya/ http://www.medactiv.ru/yguide/f/guide-f-0059.shtml
 Слайд 1
Слайд 1 Слайд 2
Слайд 2 Слайд 3
Слайд 3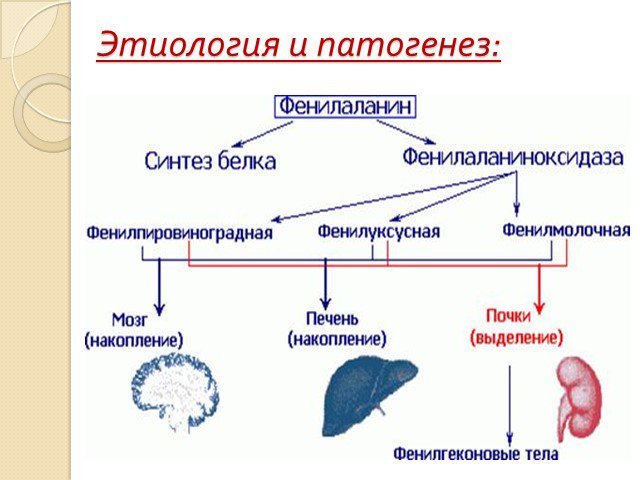 Слайд 4
Слайд 4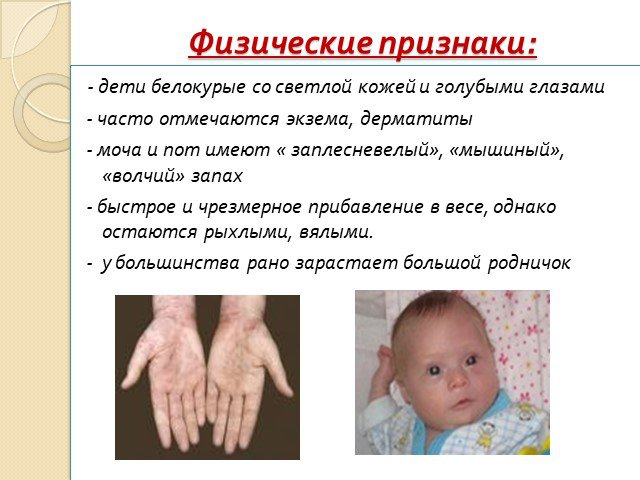 Слайд 5
Слайд 5 Слайд 6
Слайд 6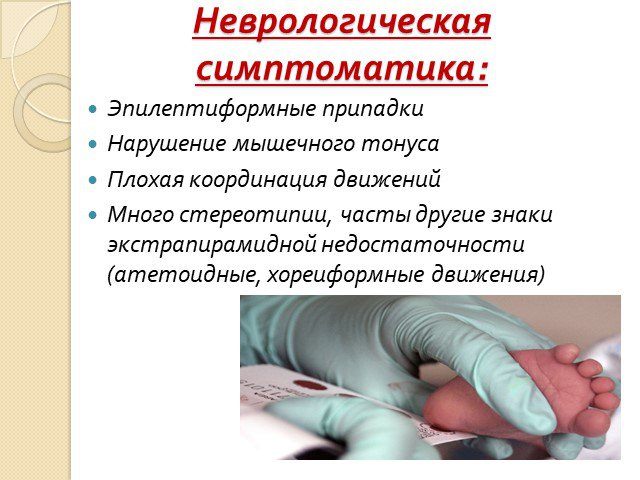 Слайд 7
Слайд 7 Слайд 8
Слайд 8 Слайд 9
Слайд 9 Слайд 10
Слайд 10 Слайд 11
Слайд 11 Слайд 12
Слайд 12 Слайд 13
Слайд 13 Слайд 14
Слайд 14 Слайд 15
Слайд 15 Слайд 16
Слайд 16 Слайд 17
Слайд 17 Слайд 18
Слайд 18 Слайд 19
Слайд 19 Слайд 20
Слайд 20








