Презентация "Синдром Марфана" по медицине – проект, доклад
 Слайд 1
Слайд 1 Слайд 2
Слайд 2 Слайд 3
Слайд 3 Слайд 4
Слайд 4 Слайд 5
Слайд 5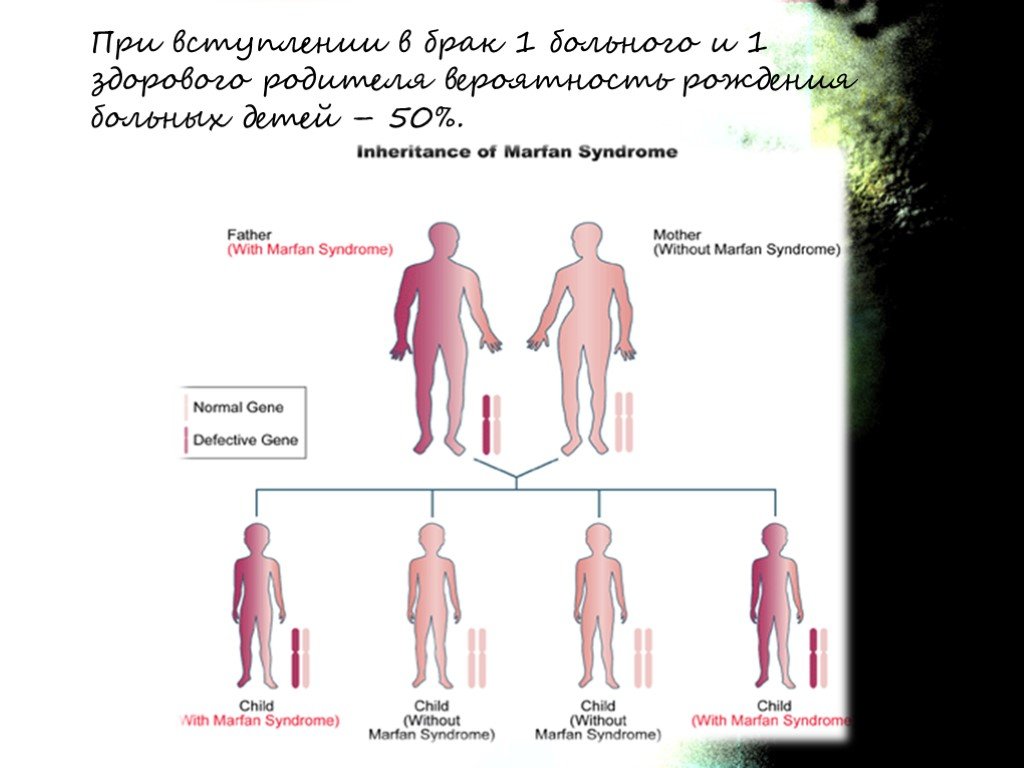 Слайд 6
Слайд 6 Слайд 7
Слайд 7 Слайд 8
Слайд 8 Слайд 9
Слайд 9 Слайд 10
Слайд 10 Слайд 11
Слайд 11 Слайд 12
Слайд 12 Слайд 13
Слайд 13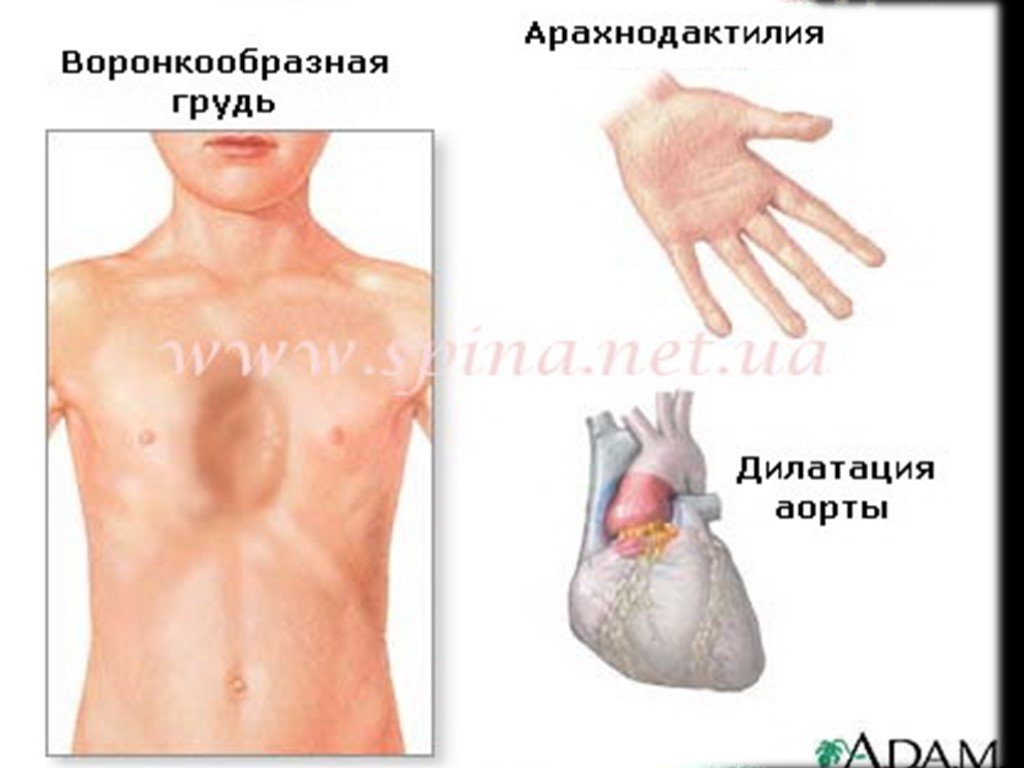 Слайд 14
Слайд 14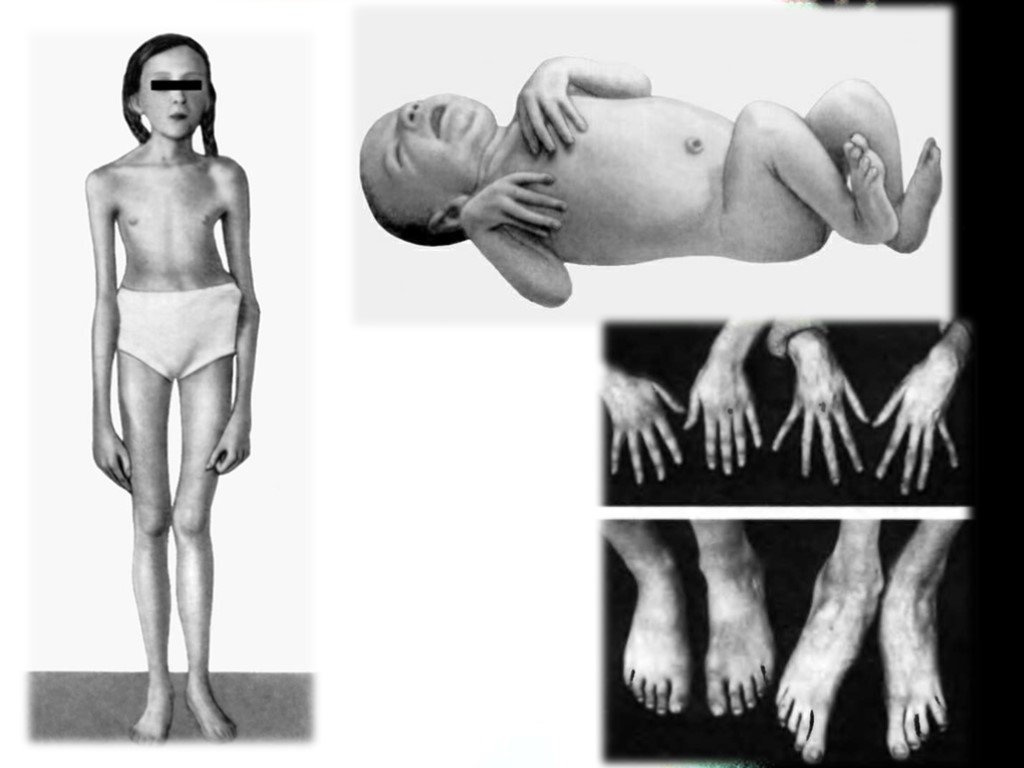 Слайд 15
Слайд 15 Слайд 16
Слайд 16 Слайд 17
Слайд 17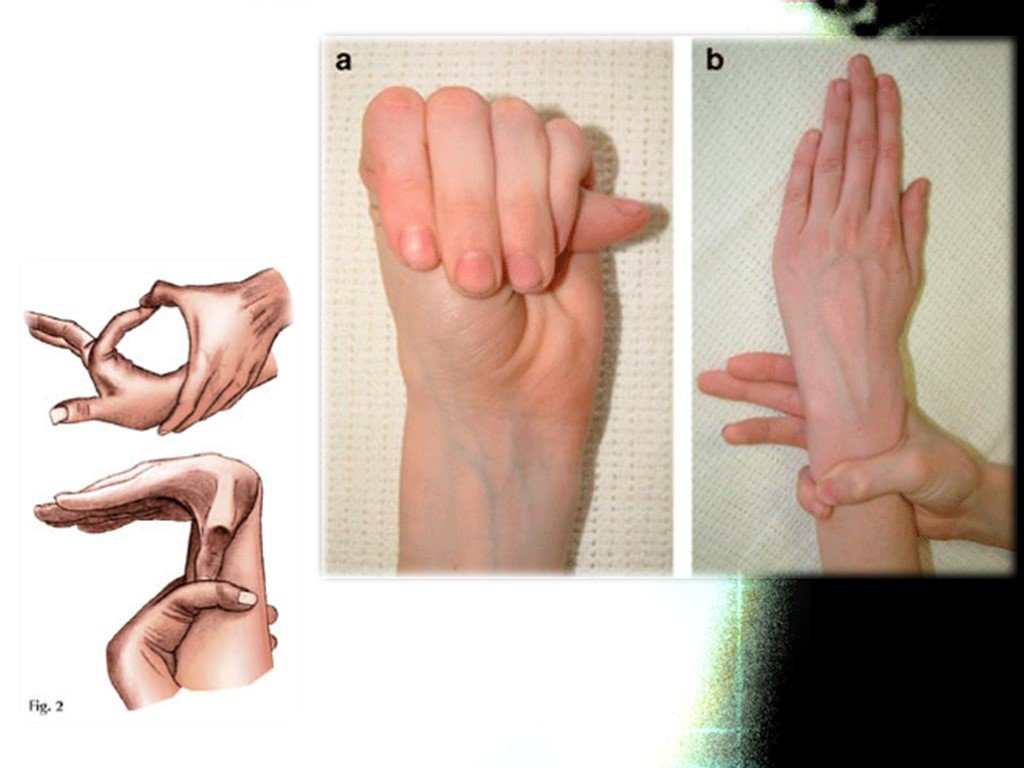 Слайд 18
Слайд 18 Слайд 19
Слайд 19 Слайд 20
Слайд 20 Слайд 21
Слайд 21 Слайд 22
Слайд 22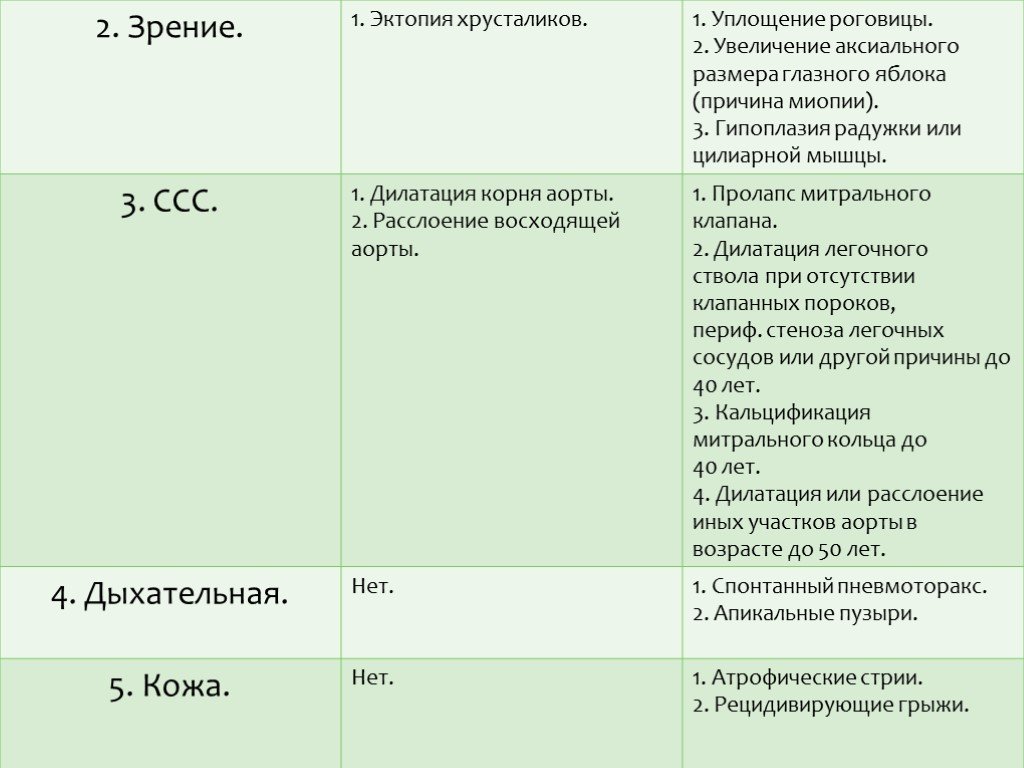 Слайд 23
Слайд 23 Слайд 24
Слайд 24 Слайд 25
Слайд 25 Слайд 26
Слайд 26 Слайд 27
Слайд 27 Слайд 28
Слайд 28 Слайд 29
Слайд 29 Слайд 30
Слайд 30Презентацию на тему "Синдром Марфана" можно скачать абсолютно бесплатно на нашем сайте. Предмет проекта: Медицина. Красочные слайды и иллюстрации помогут вам заинтересовать своих одноклассников или аудиторию. Для просмотра содержимого воспользуйтесь плеером, или если вы хотите скачать доклад - нажмите на соответствующий текст под плеером. Презентация содержит 30 слайд(ов).
Слайды презентации
Список похожих презентаций

Синдром Марфана
Происходит мутация гена фибриллина-1. Локализация гена — в длинном плече 15 хромосомы, поле 21 (15ql5-q21.3). Суть мутации — замена в белке фибриллина пролина ...
Синдром Марфана
Симптомы:. Наиболее часто встречаются долихопластический (астенический) тип, высокий рост (как правило, выше 180см) при выраженном дефиците массы ...
Синдром Леффлера
Классификация Симптомы и жалобы Причины и возбудители Патогенез Диагностика Лечение синдрома Леффлера Профилактика. Синдром Леффлера — аллергическая ...
Синдром MELAS
(англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные ...
Синдром Шихана
ЭПИДЕМИОЛОГИЯ. Это заболевание известно с конца XIX в., но только в 1937 г. H. Sheehan научно обосновал связь массивного кровотечения во время родов ...
Синдром приобретенного иммунного дефицита
Синдром приобретённого иммунного дефицита (СПИД, синдром приобретенного иммунодефицита, англ. AIDS) — состояние, развивающееся на фонеВИЧ-инфекциии ...
Синдром Патау
Трисомия 13. Частота синдрома Патау среди новорождён-ных равна 1:5000-1:7000. Характерное осложнение беременности при вынашивании плода с синдромом ...
Синдром Нунам и Шереревского-Тернера
Синдром Шерешевского-Тернера. Вперше ця хвороба як спадкова була описана в 1925 р. Н.О. Шерешевским, який вважав, що вона обумовлена недоразвиненням ...
Синдром Дауна
Что такое синдром Дауна? самая распространённая генетическая аномалия. врожденное нарушение развития, проявляющееся умственной отсталостью, нарушением ...
Синдром гипокортицизма
Надпочечники состоят из двух частей: коркового вещества – коры (на долю приходится около 80% массы железы) и мозгового вещества. В коре надпочечников ...
Синдром внешнесекреторной недостаточности поджелудочной железы. Причины. Симптомы. Диагностика. Особенности у детей
Анатомо-физиологические особенности ПЖ у детей. Поджелудочная железа имеет небольшие размеры. У новорожденного длина ее составляет 5-6 см, а к 10 ...
Синдром Бругада
В 1992 году испанские кардиологи братья Педро и Джозеф Brugada впервые описали новый клинико-электрокардиографический синдром, характеризующийся: ...
Синдром бронхиальной обструкции
Механизмы бронхиальной обструкции. Обратимый компонент: воспаление и отёк слизистой оболочки гиперплазия бокаловидных клеток гиперкриния дискриния ...
Синдром MERRF
(Myoclonic Epilepsy with Ragged Red Fibers – міоклонус епілепсія з рваними червоними м’язовими волокнами) - захворювання, обумовлене точковими мутаціями ...
Синдром малого сердечного выброса
Сплошь и рядом люди не могут договориться между собой лишь потому, что, говоря, как им кажется, об одном и том же, на самом деле толкование ими терминов ...
Синдром Дауна
Синдром Дауна – это не болезнь, а генетическое состояние, при котором в организме человека присутствует лишняя хромосома. Ребенок с нормальным развитием. ...
Синдром дауна
Задачи урока: раскрыть изменения, происходившие в правящей элите Советского Союза в 1960-80-е гг., показать, как формировалась советская партийно-государственная ...
Синдром Мендельсона. Этиопатогенез. Клиника.
Человек вышел из закусочной, за углом кто-то ударил в живот ножом, привезли на скорой, экстренная операция. Перед анестезией анестезиолог выспрашивает ...
Синдром длительного сдавливания
Синдром длительного сдавливания (СДС) - своеобразная тяжелая травма, обусловленная продолжительным сдавливанием (компрессией) мягких тканей. Он характеризуется ...
Синдром Нунан
наследственное заболевание, характеризующееся низкорослостью и аномалиями соматического развития. Синдром Нунан – это -. укороченная шея с крыловидными ...Советы как сделать хороший доклад презентации или проекта
- Постарайтесь вовлечь аудиторию в рассказ, настройте взаимодействие с аудиторией с помощью наводящих вопросов, игровой части, не бойтесь пошутить и искренне улыбнуться (где это уместно).
- Старайтесь объяснять слайд своими словами, добавлять дополнительные интересные факты, не нужно просто читать информацию со слайдов, ее аудитория может прочитать и сама.
- Не нужно перегружать слайды Вашего проекта текстовыми блоками, больше иллюстраций и минимум текста позволят лучше донести информацию и привлечь внимание. На слайде должна быть только ключевая информация, остальное лучше рассказать слушателям устно.
- Текст должен быть хорошо читаемым, иначе аудитория не сможет увидеть подаваемую информацию, будет сильно отвлекаться от рассказа, пытаясь хоть что-то разобрать, или вовсе утратит весь интерес. Для этого нужно правильно подобрать шрифт, учитывая, где и как будет происходить трансляция презентации, а также правильно подобрать сочетание фона и текста.
- Важно провести репетицию Вашего доклада, продумать, как Вы поздороваетесь с аудиторией, что скажете первым, как закончите презентацию. Все приходит с опытом.
- Правильно подберите наряд, т.к. одежда докладчика также играет большую роль в восприятии его выступления.
- Старайтесь говорить уверенно, плавно и связно.
- Старайтесь получить удовольствие от выступления, тогда Вы сможете быть более непринужденным и будете меньше волноваться.
Информация о презентации
Дата добавления:20 ноября 2018
Категория:Медицина
Содержит:30 слайд(ов)
Поделись с друзьями:
Скачать презентацию









