Презентация "Генетические синдромы" (11 класс) по биологии – проект, доклад
 Слайд 1
Слайд 1 Слайд 2
Слайд 2 Слайд 3
Слайд 3 Слайд 4
Слайд 4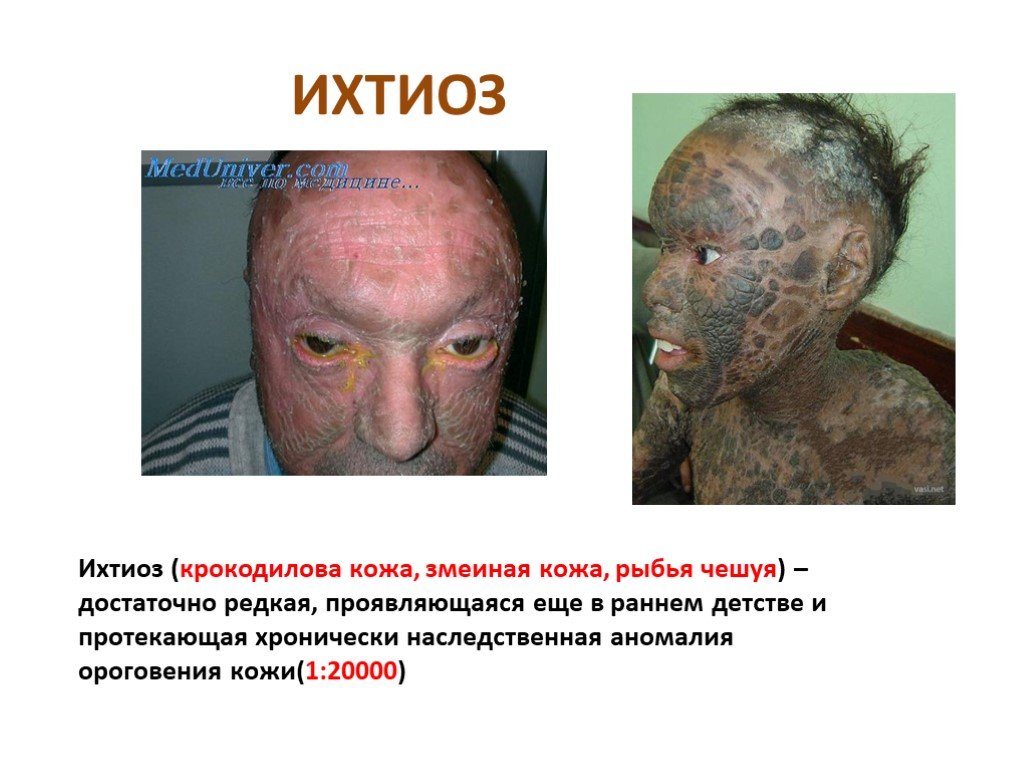 Слайд 5
Слайд 5 Слайд 6
Слайд 6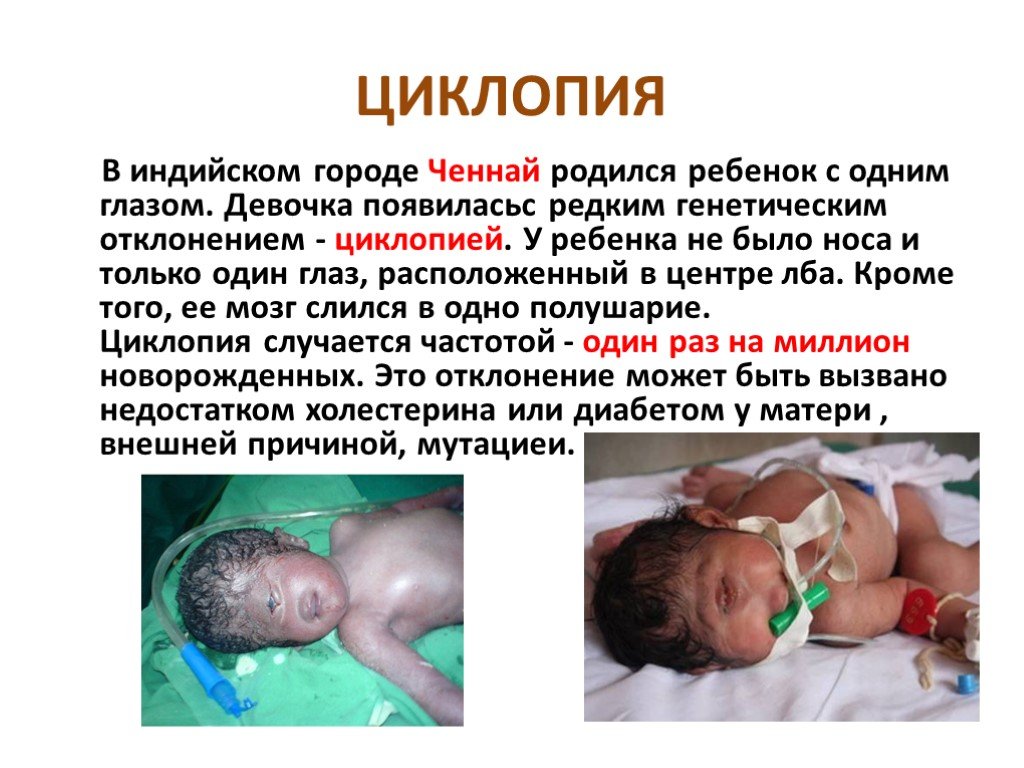 Слайд 7
Слайд 7 Слайд 8
Слайд 8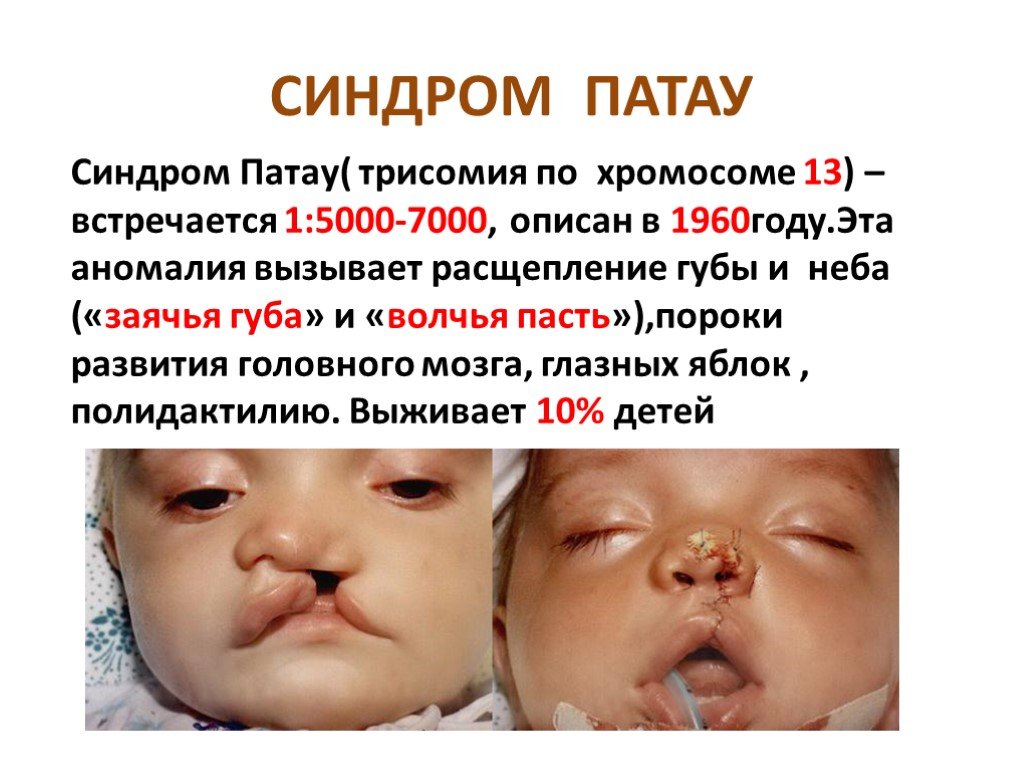 Слайд 9
Слайд 9 Слайд 10
Слайд 10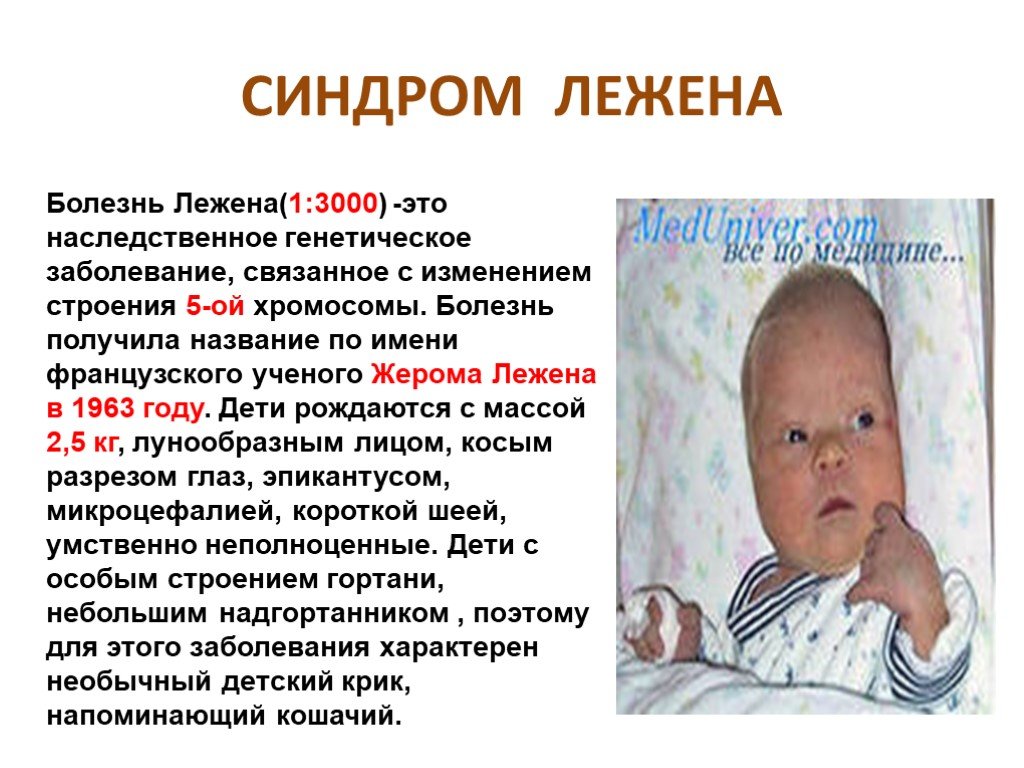 Слайд 11
Слайд 11 Слайд 12
Слайд 12 Слайд 13
Слайд 13 Слайд 14
Слайд 14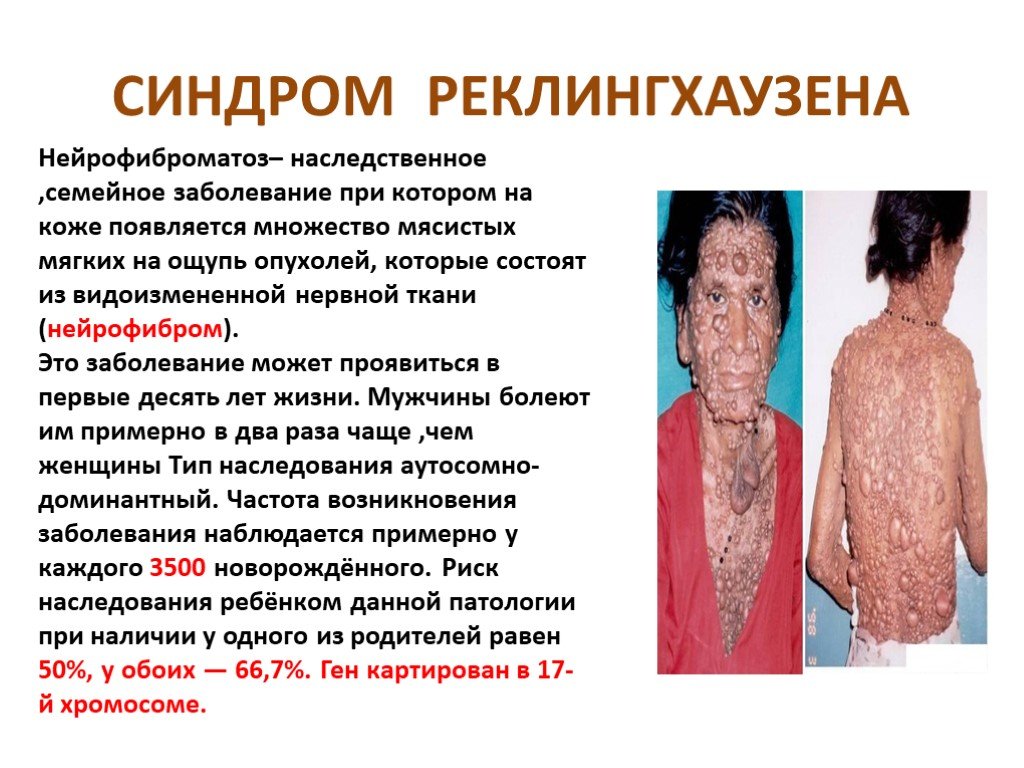 Слайд 15
Слайд 15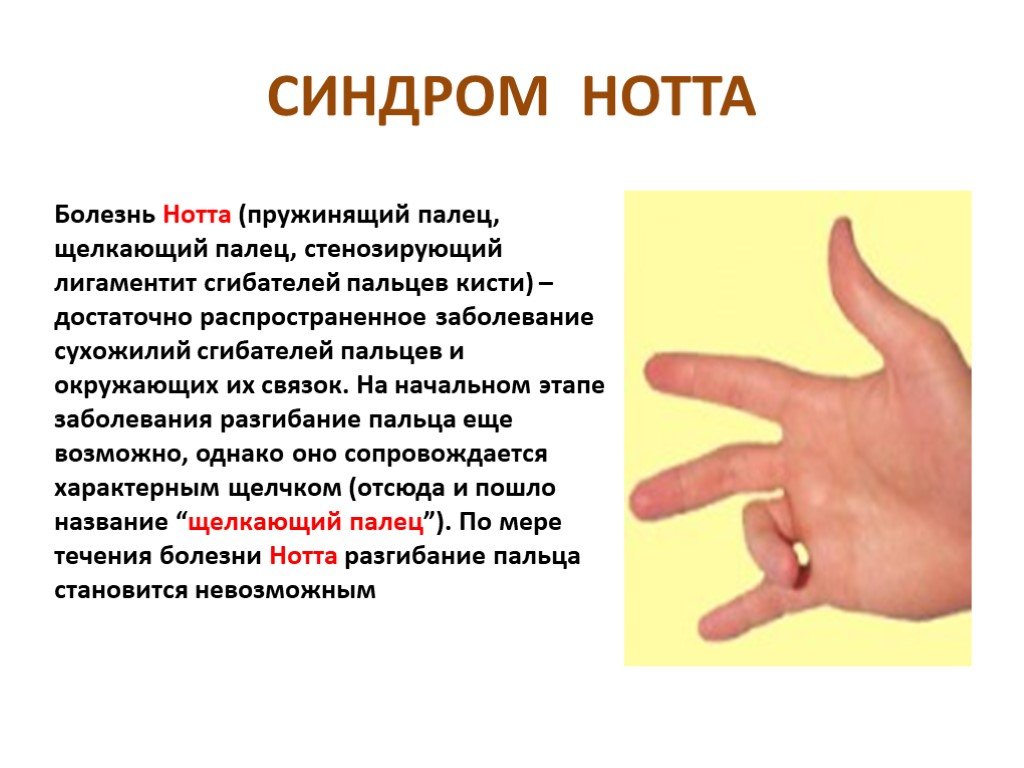 Слайд 16
Слайд 16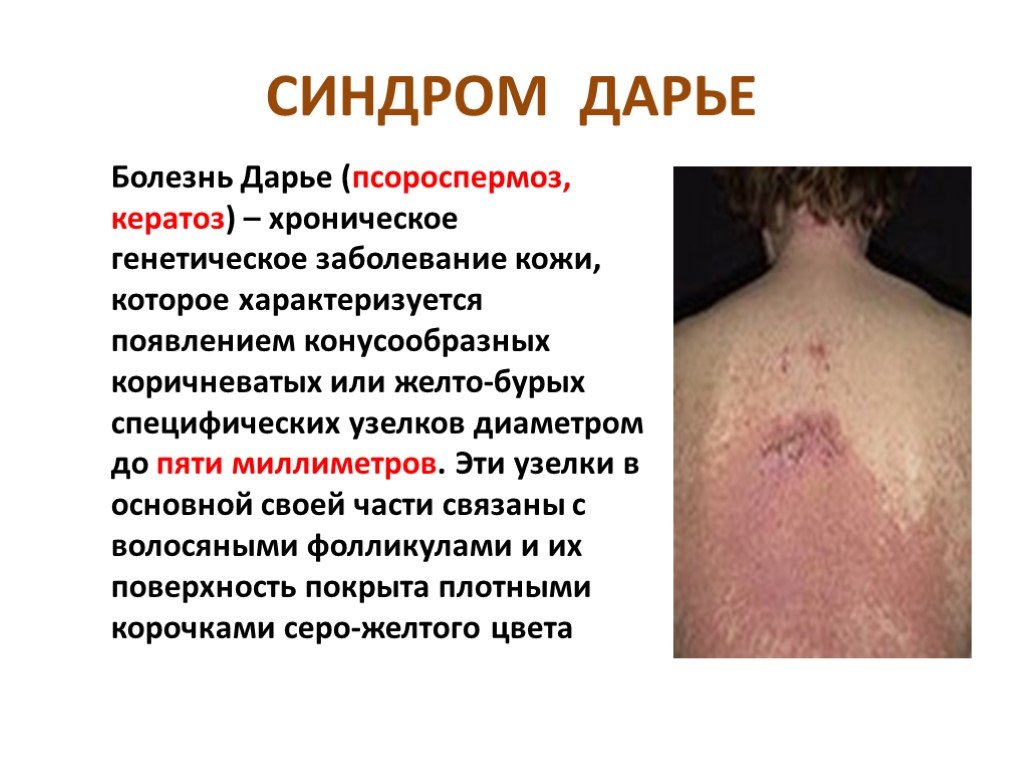 Слайд 17
Слайд 17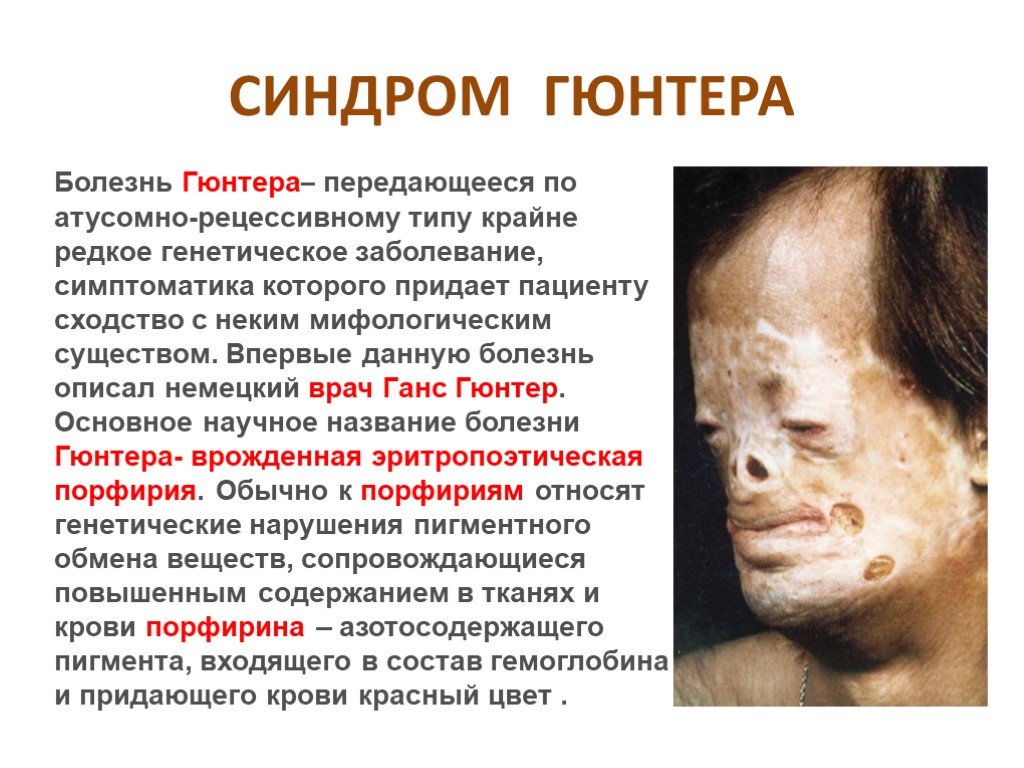 Слайд 18
Слайд 18 Слайд 19
Слайд 19 Слайд 20
Слайд 20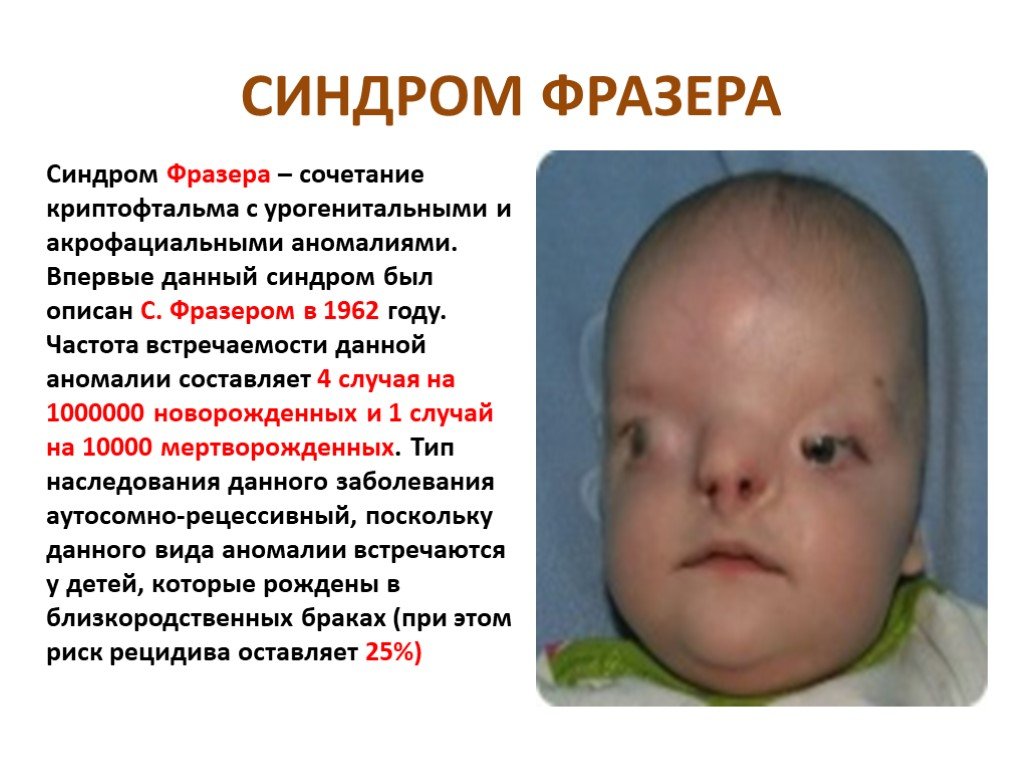 Слайд 21
Слайд 21 Слайд 22
Слайд 22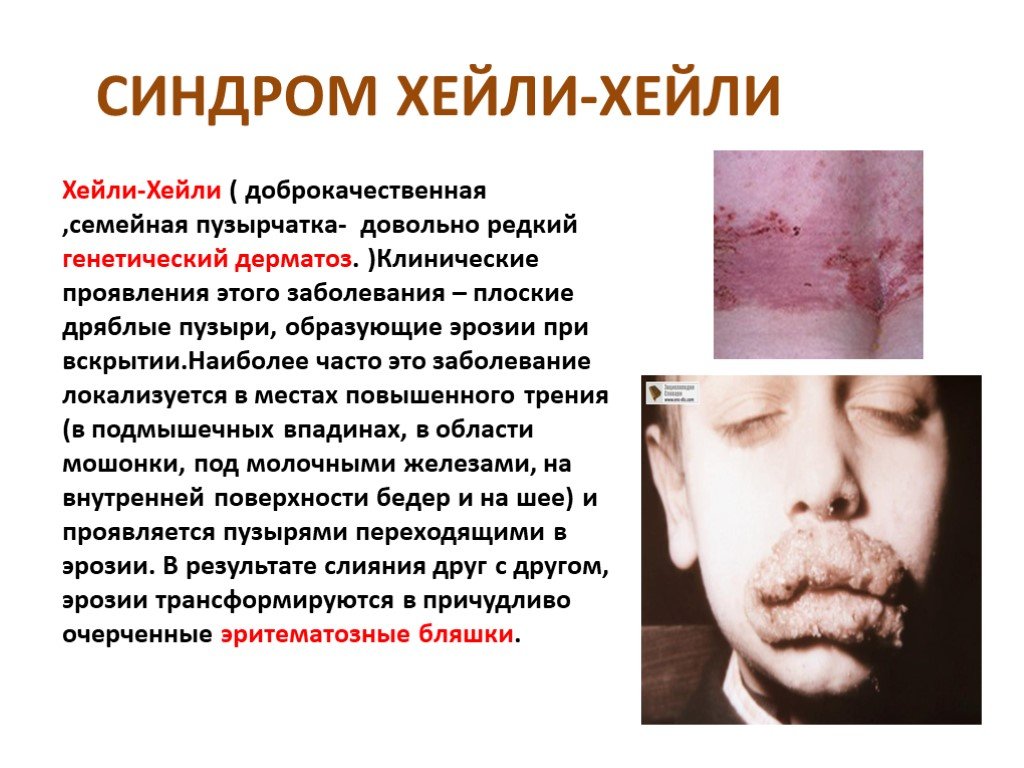 Слайд 23
Слайд 23 Слайд 24
Слайд 24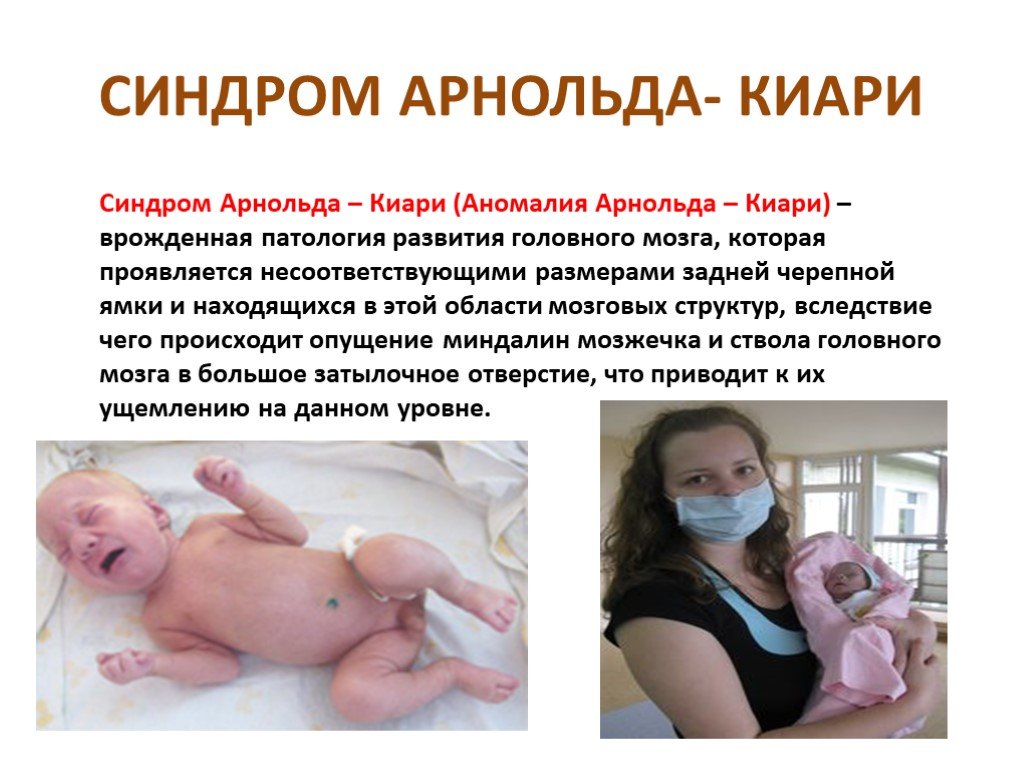 Слайд 25
Слайд 25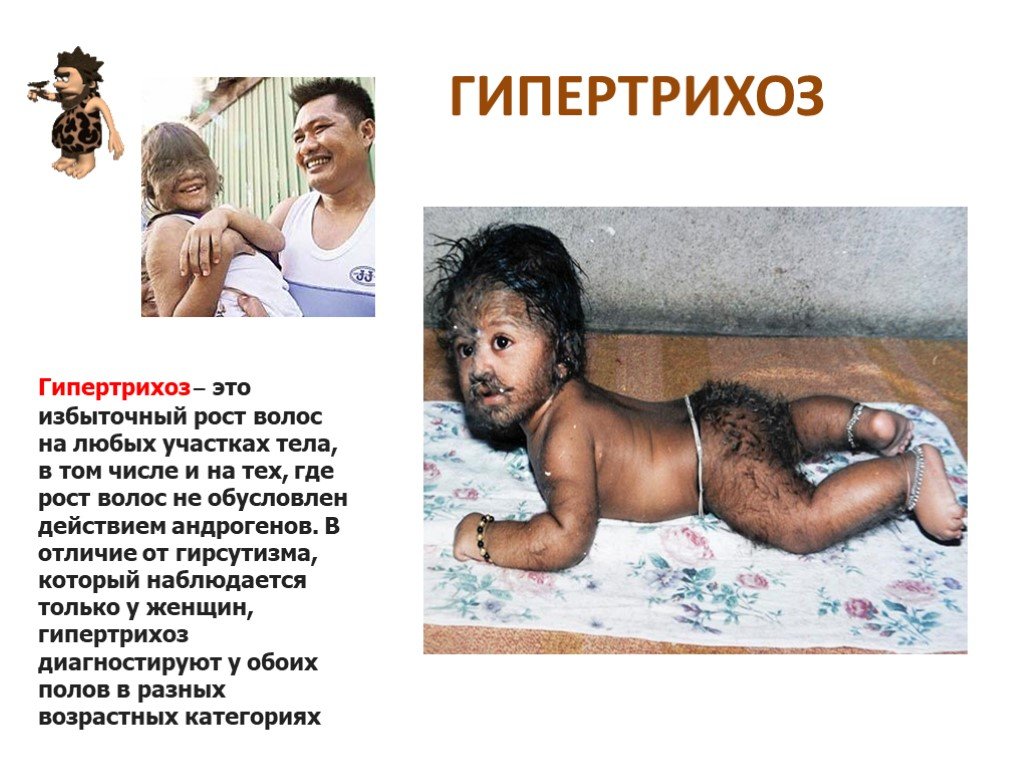 Слайд 26
Слайд 26 Слайд 27
Слайд 27 Слайд 28
Слайд 28 Слайд 29
Слайд 29 Слайд 30
Слайд 30 Слайд 31
Слайд 31 Слайд 32
Слайд 32Презентацию на тему "Генетические синдромы" (11 класс) можно скачать абсолютно бесплатно на нашем сайте. Предмет проекта: Биология. Красочные слайды и иллюстрации помогут вам заинтересовать своих одноклассников или аудиторию. Для просмотра содержимого воспользуйтесь плеером, или если вы хотите скачать доклад - нажмите на соответствующий текст под плеером. Презентация содержит 32 слайд(ов).
Слайды презентации
Список похожих презентаций

Генетические процессы в популяциях
В разных популяциях одного вида частота мутантных генов неодинакова. Это, в основном, обусловлено тем, что популяции обитают в неодинаковых условиях ...
Генетические факторы, их воздействие на здоровье
Методы предупреждения наследственных заболеваний. Основной метод предупреждения наследственны заболеваний заключается в медико –генетическом консультировании ...
Генетические основы эволюции
Ч. Дарвин различал определенную (ненаследственную) и неопределенную (наследственную) изменчивость. В настоящее время такое разделение справедливо ...
Генетические особенности наследования групп крови
иммуногематология с успехом используется в судебной медицине при спорах об отцовстве, материнстве и в случае потери детей в раннем возрасте. определение ...
Генетические основы селекции организмов
СЕЛЕКЦИЯ -. это наука, изучающая биологические основы и методы создания или улучшения пород животных, сортов растений и штаммов микроорганизмов. Селекционер ...
Генетические опыты менделя
генетические опыты Менделя. 1.Сформировать умение решать генетические задачи. 2.Добиться понимания универсального характера законов наследования. ...
Генетические опыты менделя
генетические опыты Менделя. 1.Сформировать умение решать генетические задачи. 2.Добиться понимания универсального характера законов наследования. ...
Генетические аспекты селекции лошадей
Систематическое положение лошади. Царство: Животные (Zoa, Animalia) Тип: Хордовые (Chordata) Подтип: Позвоночные (Vertebrata) Класс: Млекопитающие ...
Генетические алгоритмы
Понятие генетического алгоритма. Генети́ческий алгори́тм (англ. genetic algorithm) — это эвристический алгоритм поиска, применяемый для решения задач ...
Иммунитет. профилактика инфекционных заболеваний
Проверь себя ! 1-б; 2-г; 3-г; 4-б; 5-в; 6-г; 7-б ; 8-в; 9-в. Профилактика инфекционных заболеваний. «Измеряй всё доступное измерению и делай не доступное ...
Нарушения нервной деятельности и профилактика заболеваний
Цели и задачи. - познакомимся с нарушениями деятельности нервной системы и факторами вредящими здоровью; влияние алкоголя, курения, наркотических ...
Общая характеристика и особенности внешнего строения млекопитающих
Цель урока:. познакомится с общей характеристикой и особенностями внешнего строения млекопитающих как высокоорганизованных животных. Задачи урока:. ...
Биология и генетика пола
Пол – комплекс морфологических, физиологических, биохимических и поведенческих признаков организма, которые обеспечивают процесс воспроизведения себе ...
Микробиология – как наука. история микробиологии
МИКРОБИОЛОГИЯ – КОМПЛЕКС БИОЛОГИЧЕСКИХ НАУК, ИЗУЧАЮЩИХ МОРФОЛОГИЮ, ФИЗИОЛОГИЮ, ГЕНЕТИКУ, ЭКОЛОГИЮ И ЭВОЛЮЦИЮ МИКРООРГАНИЗМОВ. БАКТЕРИОЛОГИЯ, ВИРУСОЛОГИЯ, ...
Генетика: история развития науки. Основные понятия
ЗАДАЧИ УРОКА:. Познакомиться с наукой «генетика», ее историей и достижениями. Определить цели и задачи генетики в современном мире. Показать роль ...
Краткая история эмбриологии
Краткая история эмбриологии. Древняя Греция: Гиппократ vs Аристотель. Аристотель (384-322 до н.э.). «О возникновении животных» (начало сравнительной ...
Красная книга ее роль и история
Наша планета – живая, её населяют около 2 млн известных видов живых организмов. Однако уже в конце XIX века стало понятно, что виды вымирают, и все ...
Клетка история изучения. клеточная теория
Уровни организации живого. Молекулярно-генетический Клеточный Тканевый Органный Организменный Популяционно-видовой Биоценотический Биосферный. Клетка ...
Ангина и её лечение
Ангина. Ангина – заболевание инфекционное, передающееся воздушно-капельным путем. Чаще всего возбудителем ангины становятся стрептококки. Болезнь ...
Общая характеристика грибов. шляпочные грибы
ГРИБЫ. Подосиновики, маслята, рыжики, грузди, валуи. Мухоморы, поганки. Насчитывается около 100 тыс. видов. Не содержат хлорофилла. Встречаются повсеместно. ...Конспекты
Генетические символы . Решение задач на моногибридное скрещивание
План конспект урока № 37 Дата проведения –21.01.2013. Класс -9 «а». Тема :Генетические символы . Решение задач на моногибридное скрещивание. . ...Советы как сделать хороший доклад презентации или проекта
- Постарайтесь вовлечь аудиторию в рассказ, настройте взаимодействие с аудиторией с помощью наводящих вопросов, игровой части, не бойтесь пошутить и искренне улыбнуться (где это уместно).
- Старайтесь объяснять слайд своими словами, добавлять дополнительные интересные факты, не нужно просто читать информацию со слайдов, ее аудитория может прочитать и сама.
- Не нужно перегружать слайды Вашего проекта текстовыми блоками, больше иллюстраций и минимум текста позволят лучше донести информацию и привлечь внимание. На слайде должна быть только ключевая информация, остальное лучше рассказать слушателям устно.
- Текст должен быть хорошо читаемым, иначе аудитория не сможет увидеть подаваемую информацию, будет сильно отвлекаться от рассказа, пытаясь хоть что-то разобрать, или вовсе утратит весь интерес. Для этого нужно правильно подобрать шрифт, учитывая, где и как будет происходить трансляция презентации, а также правильно подобрать сочетание фона и текста.
- Важно провести репетицию Вашего доклада, продумать, как Вы поздороваетесь с аудиторией, что скажете первым, как закончите презентацию. Все приходит с опытом.
- Правильно подберите наряд, т.к. одежда докладчика также играет большую роль в восприятии его выступления.
- Старайтесь говорить уверенно, плавно и связно.
- Старайтесь получить удовольствие от выступления, тогда Вы сможете быть более непринужденным и будете меньше волноваться.
Информация о презентации
Дата добавления:11 ноября 2018
Категория:Биология
Содержит:32 слайд(ов)
Поделись с друзьями:
Скачать презентацию









